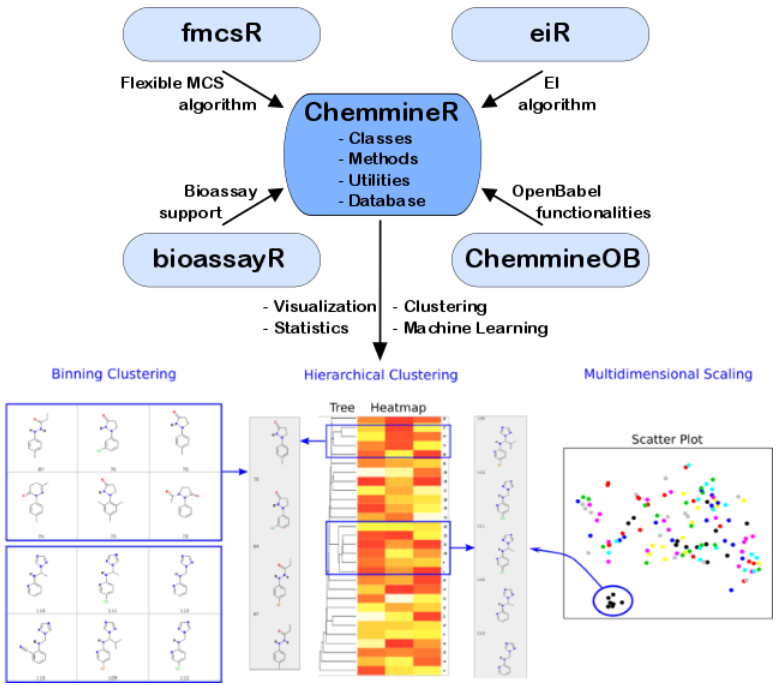
ChemmineR是使用R语言实现化合物基础操作的工具包,现根据其官方文档学习其主要用法如下:
1
2
3
4
5
6
|
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("ChemmineR")
library("ChemmineR")
# library("ChemmineOB")
|
1. SDFset格式#
- ChemmineR基础操作是围绕SDFset对象展开的,其表示多个SDF格式的化合物集合
1
2
3
4
5
6
7
8
9
|
data(sdfsample)
sdfset = sdfsample
# valid <- validSDF(sdfset)
# sdfset <- sdfset[valid]
class(sdfset) # SDFset
length(sdfset) # 100
c(sdfset[1:4], sdfset[5:8]) # 合并
sdfset[1:4] # 子集
|
- 每个SDFset集合是由单个SDF对象组成的,主要由4部分构成
- <<header» : 化合物id等基本信息
- <<atomblock» : 原子信息,<<bondblock»: 键信息
- <<datablock» : 化合物的属性/其它注释信息
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
sdfset[[1]]
as(sdfset[[1]], "list")
## ID
cid(sdfset[1:2]) # slot ID
sdfid(sdfset[1:2]) # header ID
cid(sdfset) = sdfid(sdfset)
## Component
header(sdfset[[1]]) # character
atomblock(sdfset[[1]]) # matrix
bondblock(sdfset[[1]]) # matrix
datablock(sdfset[[1]]) # character
blockmatrix = datablock2ma(datablock(sdfset[1:2]))
|
补充:ChemmineR提供一些函数可计算化合物的基本属性信息,例如分子量等。此外ChemmineOB也可以实现类似功能。
1
2
3
4
5
6
7
8
9
10
11
12
13
|
## ChemmineR
propma <- data.frame(MF=MF(sdfset, addH=FALSE), MW=MW(sdfset, addH=FALSE),
Ncharges=sapply(bonds(sdfset, type="charge"), length),
atomcountMA(sdfset, addH=FALSE),
groups(sdfset, type="countMA"),
rings(sdfset, upper=6, type="count", arom=TRUE))
## based on ChemmineOB
propma = propOB(sdfset[1])
colnames(propma)
# [1] "cansmi" "cansmiNS" "formula" "title" "InChI" "HBA1" "HBA2"
# [8] "HBD" "logP" "MR" "MW" "nF" "TPSA"
# datablock(sdfset) <- cbind(datablock(sdfset), propma)
|
2. 格式转换(smi)#
1
2
3
|
smiles <- sdf2smiles(sdfset[1:2])
class(smiles)
cid(smiles)
|
- smiles string → SMIset/SDFset
1
2
3
|
sdf <- smiles2sdf("CC(=O)OC1=CC=CC=C1C(=O)O")
as(as.character(smiles), "SMIset")
|
- 此外ChemmineOB支持更多化合物格式间的转换
1
2
3
4
5
6
7
8
9
|
## R object
convertFormat("smi","sdf","CC(=O)OC1=CC=CC=C1C(=O)O")
convertFormat("smi","mol2","CC(=O)OC1=CC=CC=C1C(=O)O")
## File
## 将smi文件(第一列smi,第二列optional ID)转换为sdf文件
convertFormatFile("smi","sdf","example.smi","example.sdf")
# 可参见obabel的笔记了解其支持的全部格式
|
3. 导出与读入#
1
2
3
4
5
6
7
8
9
10
11
12
|
# 一般导出
write.SDF(sdfset[1:4], file="sub.sdf")
# 分批导出
write.SDFsplit(x=sdfstr, filetag="myfile", nmol=10)
# 对于大量分子,可保存为二进制rda,减少文件大小
save(sdfstr, file="sdfstr.rda")
# 读入为SDFset
subsdfset <- read.SDFset("sub.sdf")
|
1
2
3
4
5
|
data(smisample)
smiset <- smisample
write.SMI(smiset[1:4], file="sub.smi")
smiset <- read.SMIset("sub.smi")
|
4. AP/FP特征#
- AP表示原子对特征(Atom Pair Descriptors)。APset 表示若干化合物的AP特征集合
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
sdfset = sdfsample
ap <- sdf2ap(sdfset[[1]]) # single
apset <- sdf2ap(sdfset) # many(common)
## 计算特定两两相似度
cmp.similarity(apset[1],apset[2])
# [1] 0.2637037
## 批量计算
cmp.search(apset,apset[1], type=3, cutoff = 0.3, quiet=TRUE)
# index cid scores
# 1 1 CMP1 1.0000000
# 2 96 CMP96 0.3516643
# 3 67 CMP67 0.3117569
# 4 88 CMP88 0.3094629
# 5 15 CMP15 0.3010753
|
- FP表示指纹编码特征(Fingerprints)。FPset表示若干化合物的FP特征集合。
(1)ChemmineR内置了一种根据APset计算FP的函数
1
2
3
4
5
|
?desc2fp
fpset <- desc2fp(apset, descnames=1024, type="FPset")
fpma <- desc2fp(apset, descnames=512, type="matrix")
fpch <- desc2fp(apset, descnames=64, type="character")
|
(2)ChemmineOB支持计算化合物的ECFP、SMARTs等指纹类型(详见obabel笔记)
1
2
|
fpset <-fingerprintOB(sdfset,"FP2")
fpset <-fingerprintOB(sdfset,"ECFP10")
|
(3)计算化合物间FP的相似度
1
|
fpSim(fpset[1], fpset, method="Tanimoto")
|
5. Pubchem接口#
- fingerprintR提供简单的pubchem api调用,用于特征场景的查询
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
# (1) PID → SDFset
sdfset_01 <- pubchemCidToSDF(c(111,123))
# (2) inchikeys → SDFset
inchikeys <- c(
"ZFUYDSOHVJVQNB-FZERPYLPSA-N",
"KONGRWVLXLWGDV-BYGOPZEFSA-N",
"AANKDJLVHZQCFG-WLIQWNBFSA-N",
"SNFRINMTRPQQLE-JQWAAABSSA-N"
)
inchikey_query <- pubchemInchikey2sdf(inchikeys)
sdfset_02 = inchikey_query$sdf_set
sdfid(sdfset_02)
# [1] "162880185" "11276107" "442060"
|