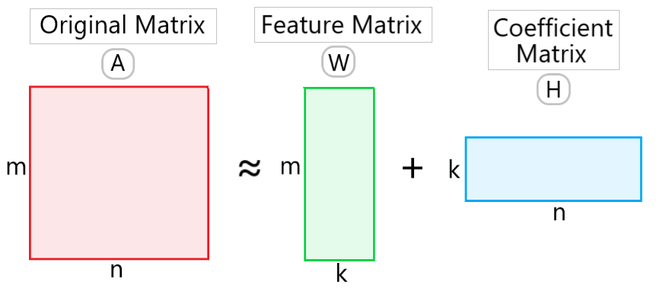
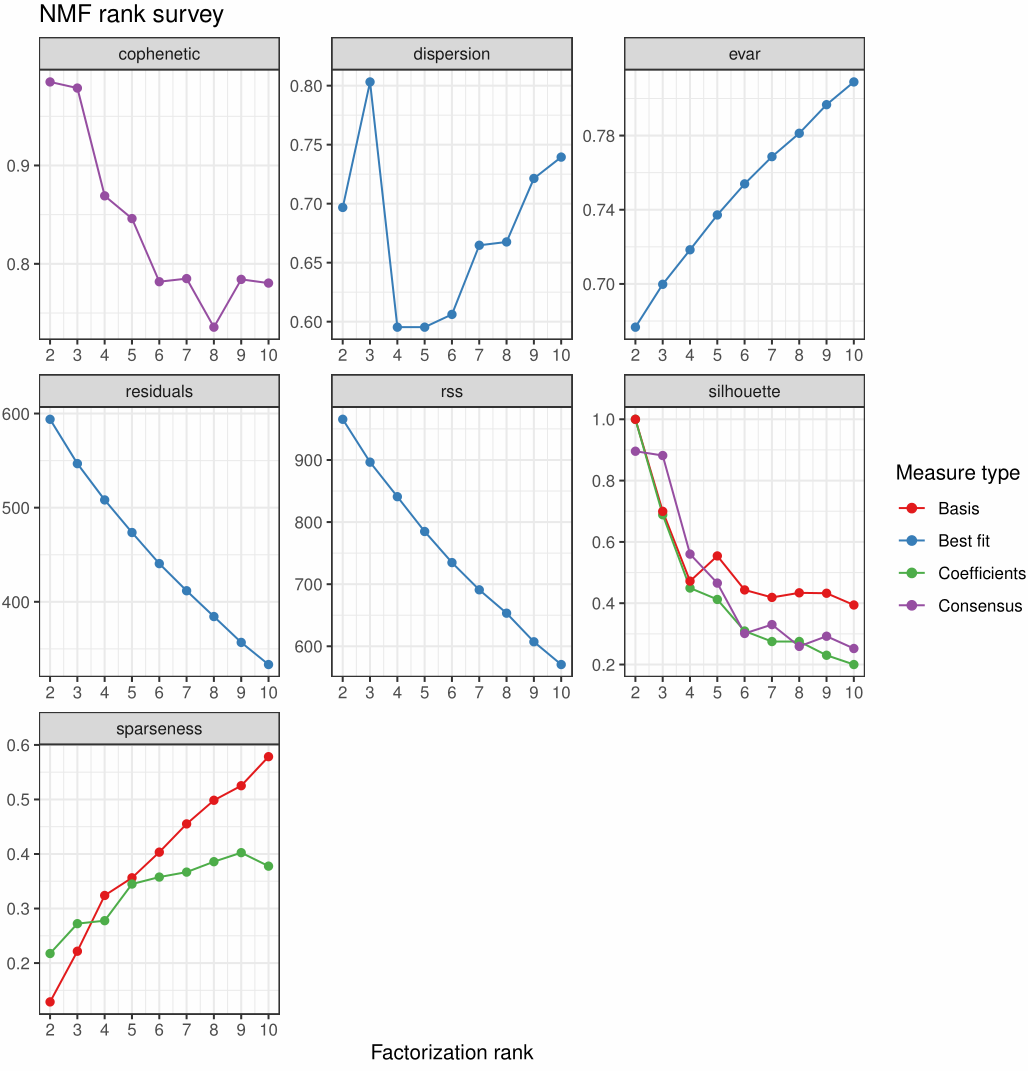
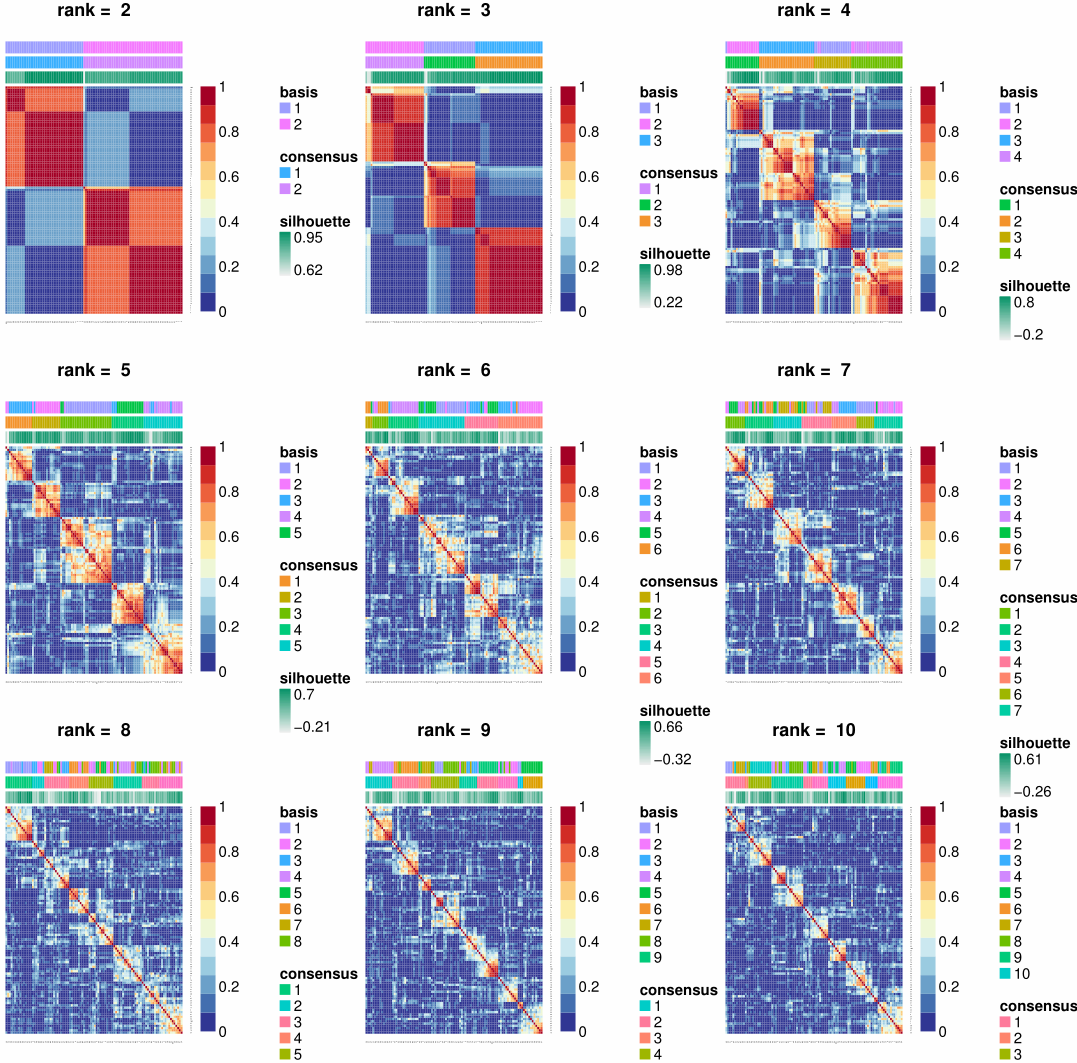
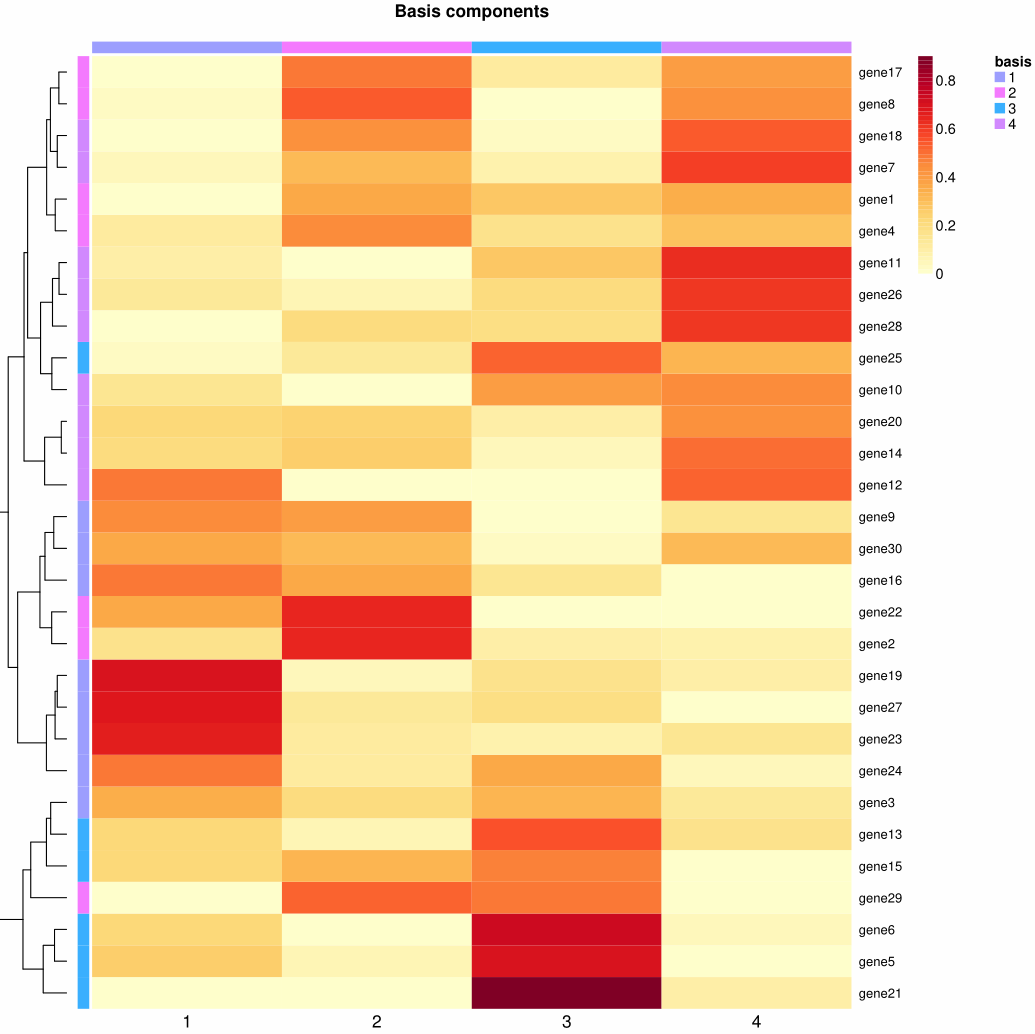
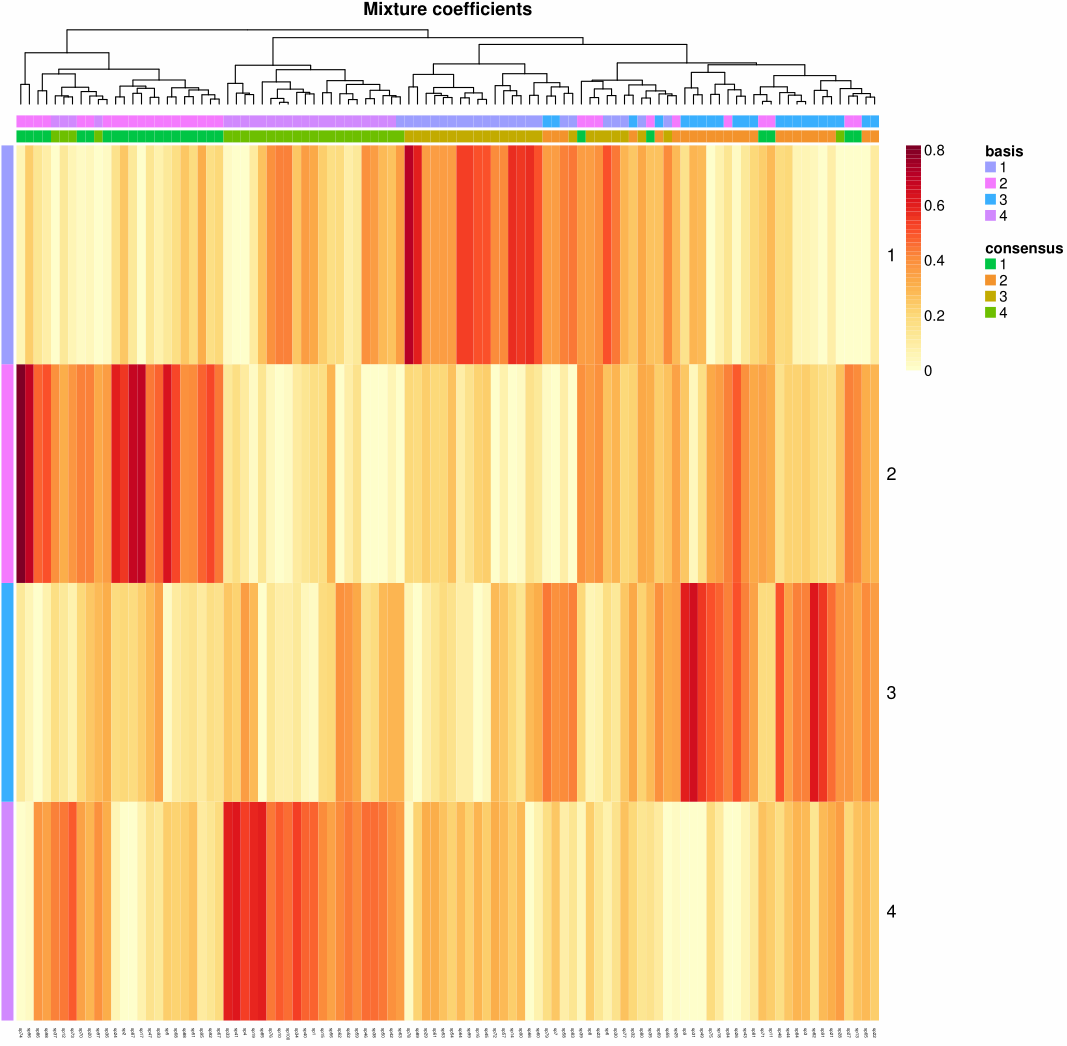
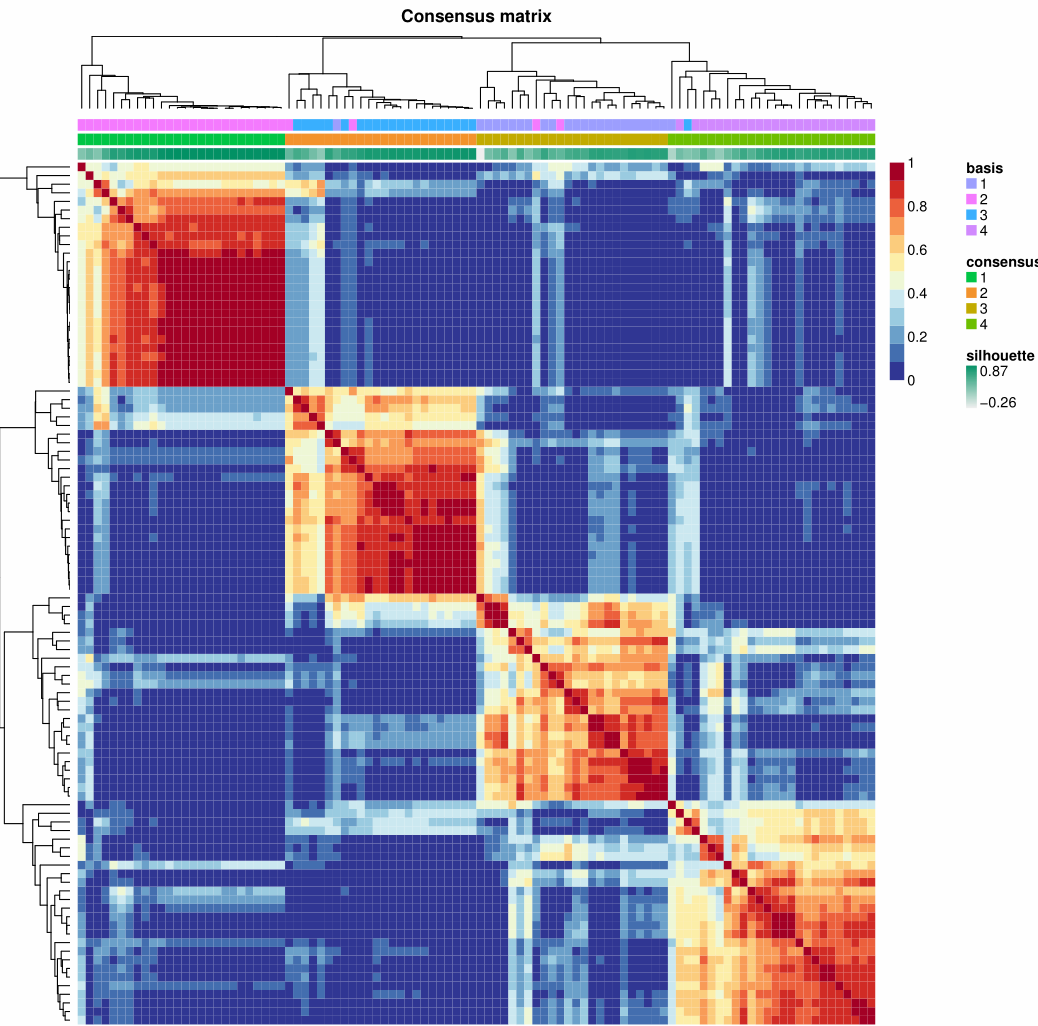
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
|
library(NMF)
# 支持算法:c("brunet","lee","ls-nmf","nsNMF","offset","pe-nmf","snmf/r","snmf/l")
nmf_model <- nmf(exp_mat, rank = 3, method = "brunet", seed = 42)
# 获取基矩阵(基因--细胞)
basis_matrix <- basis(nmf_model)
dim(basis_matrix)
# [1] 30 3
head(basis_matrix)
# [,1] [,2] [,3]
# gene1 1.7436283 0.7842223 3.0718832
# gene2 1.0542018 0.9599996 2.8275129
# gene3 2.7498596 1.4277110 1.0828163
# gene4 0.8918389 1.8432274 2.7861665
# gene5 4.2662810 0.8883752 0.3467359
# gene6 5.8054591 0.6420813 0.2967837
# 获取系数矩阵(细胞--样本)
coef_matrix <- coef(nmf_model)
dim(coef_matrix)
# [1] 3 100
head(coef_matrix[,1:3])
# sp1 sp2 sp3
# [1,] 0.002897117 0.02866644 0.02137146
# [2,] 0.131645724 0.14692969 0.04635707
# [3,] 0.022945508 0.05598917 0.11973042
# [4,] 0.169223269 0.06491003 0.13558182
# [5,] 0.088389864 0.10569255 0.12587923
|