1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
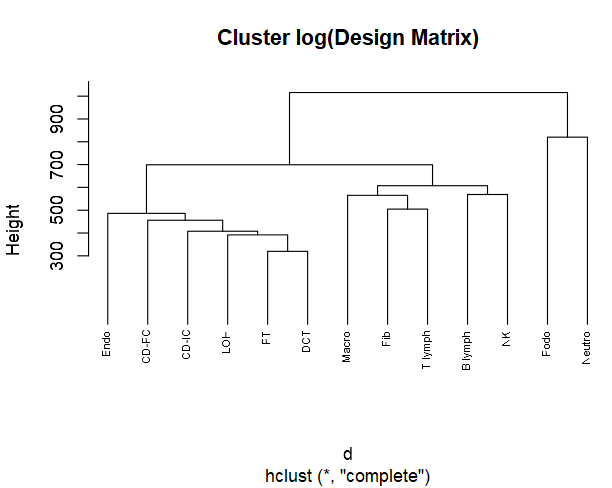
d <- dist(t(log(Mousesub.basis$Disgn.mtx + 1e-6)), method = "euclidean")
# d <- dist(t(log(Mousesub.basis$M.theta + 1e-8)), method = "euclidean")
hc1 <- hclust(d, method = "complete" )
plot(hc1, cex = 0.6, hang = -1, main = 'Cluster log(Design Matrix)')
clusters.type = list(C1 = 'Neutro',
C2 = 'Podo',
C3 = c('Endo', 'CD-PC', 'LOH', 'CD-IC', 'DCT', 'PT'),
C4 = c('Macro', 'Fib', 'B lymph', 'NK', 'T lymph'))
# 添加到单细胞数据中
cl.type = as.character(Mousesub.sce$cellType)
for(cl in 1:length(clusters.type)){
cl.type[cl.type %in% clusters.type[[cl]]] = names(clusters.type)[cl]
}
Mousesub.sce$clusterType = factor(cl.type,
levels = c(names(clusters.type), 'CD-Trans', 'Novel1', 'Novel2'))
|