xCell#
xCell包#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
devtools::install_github('dviraran/xCell')
library(xCell)
data("xCell.data")
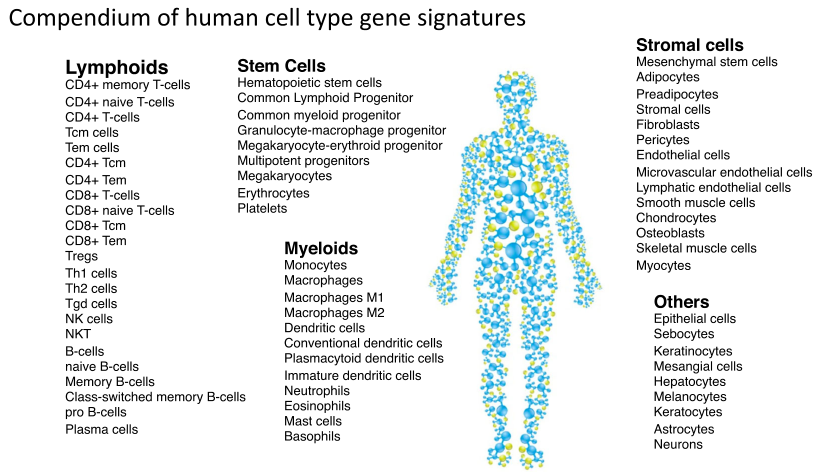
##查看支持的64种细胞类型,同下图
colnames(xCell.data$spill$K)
##预测函数的关键参数解释
?xCellAnalysis()
# expr = 交代表达矩阵;
##如果是array,不需要额外标准化;如果是RNAseq,需要TPM/FPKM/TPM。
##对于基因ID格式需要是symbol格式。
# rnaseq = TRUE 数据是否为RNAseq数据,如果是芯片数据设置为FALSE
# cell.types.use = NULL 提供一个字符串,说明想要预测64种细胞中的哪些细胞类型
# parallel.sz = 4 调用的线程数,默认为4
|
NOTE:
(1) 由于xCell支持64种细胞类型,分析前根据先验知识判断预期有哪些细胞类型再分析,会提高数据结果可靠度性。
(2) xCell的结果适用于同一细胞类型在不同样本间的含量差异比较,不适用于同一样本的不同细胞类型组成分析。
示例分析#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
|
##(1)加载包
library(xCell)
data("xCell.data")
##(2)示例表达数据:xCell内嵌的array数据
sdy = readRDS('sdy420.rds')
expr = sdy$expr
dim(expr) #104个样本的12027个样本
# [1] 12027 104
expr[1:4,1:4]
# SUB137169 SUB137172 SUB137208 SUB137209
# A1CF 6.3480 6.1807 5.9556 6.3678
# A2LD1 6.8177 8.6354 7.2744 6.8724
# A2M 6.7169 6.9159 7.3686 6.7054
# A2ML1 7.8906 9.8874 10.5970 9.0120
##(3)选取预期的细胞类型用于预测
cell.types.use = intersect(colnames(xCell.data$spill$K),
rownames(sdy$fcs))
cell.types.use
# [1] "B-cells" "CD4+ naive T-cells" "CD4+ T-cells"
# [4] "CD4+ Tcm" "CD4+ Tem" "CD8+ naive T-cells"
# [7] "CD8+ T-cells" "CD8+ Tcm" "CD8+ Tem"
# [10] "Memory B-cells" "Monocytes" "naive B-cells"
# [13] "NK cells" "NKT" "Plasma cells"
# [16] "pro B-cells" "Tgd cells" "Tregs"
##(4) xCellAnalysis分析
scores = xCellAnalysis(expr, rnaseq=F,
cell.types.use = cell.types.use)
#得到104样本的18种细胞类型的含量
dim(scores)
# [1] 18 104
scores[1:4,1:4]
# SUB137169 SUB137172 SUB137208 SUB137209
# B-cells 0.11282951 1.689912e-01 0.15363674 0.11287388
# CD4+ naive T-cells 0.06465254 0.000000e+00 0.09620357 0.07204639
# CD4+ T-cells 0.18315694 8.676322e-02 0.14341114 0.14930520
# CD4+ Tcm 0.06477823 4.463612e-18 0.02393895 0.01463585
|
CIBERSOFT#
CIBERSORT脚本#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
|
source("CIBERSORT.R")
# 使用CIBERSORT()函数预测免疫浸润细胞比例
# 其中涉及两个关键参数:
##(1) sig_matrix:参考细胞类型特征表达矩阵文件名,官方提供了LM22.txt
LM22 = data.table::fread("LM22.txt")
dim(LM22)
# [1] 547 23
LM22[1:4,1:4]
# Gene symbol B cells naive B cells memory Plasma cells
# 1: ABCB4 555.71345 10.74423 7.225819
# 2: ABCB9 15.60354 22.09479 653.392328
# 3: ACAP1 215.30595 321.62102 38.616872
# 4: ACHE 15.11795 16.64885 22.123737
colnames(LM22) #所覆盖的22种免疫细胞
# [1] "Gene symbol" "B cells naive" "B cells memory"
# [4] "Plasma cells" "T cells CD8" "T cells CD4 naive"
# [7] "T cells CD4 memory resting" "T cells CD4 memory activated" "T cells follicular helper"
# [10] "T cells regulatory (Tregs)" "T cells gamma delta" "NK cells resting"
# [13] "NK cells activated" "Monocytes" "Macrophages M0"
# [16] "Macrophages M1" "Macrophages M2" "Dendritic cells resting"
# [19] "Dendritic cells activated" "Mast cells resting" "Mast cells activated"
# [22] "Eosinophils" "Neutrophils"
##(2) mixture_file:待预测细胞组成的表达矩阵文件名
# 如果是array,不需要额外标准化;如果是RNAseq,需要TPM/FPKM/TPM。
# 对于基因ID格式需要是symbol格式。
# 将符合上述两个条件的表达矩阵储存为Tab分割的TXT文件(行名转为第一列数据,列名为“symbol”,不储存行名)
|

示例分析#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
|
##(1)加载脚本
source("CIBERSORT1.03.R")
##(2)示例表达数据:仍使用上面xCell内嵌的array数据
sdy = readRDS('sdy420.rds')
expr = sdy$expr
dim(expr) #104个样本的12027个样本
# [1] 12027 104
expr[1:4,1:4]
expr2 = tibble::rownames_to_column(expr, "symbol")
expr2[1:4,1:4]
# symbol SUB137169 SUB137172 SUB137208
# 1 A1CF 6.3480 6.1807 5.9556
# 2 A2LD1 6.8177 8.6354 7.2744
# 3 A2M 6.7169 6.9159 7.3686
# 4 A2ML1 7.8906 9.8874 10.5970
write.table(expr2, row.names = F,quote = F,sep = "\t",
file = "exp_array.txt")
##(3)官方提供的参考细胞类型特征表达矩阵文件:"LM22.txt"
##(4)CIBERSORT预测
results = CIBERSORT(sig_matrix = "LM22.txt",
mixture_file = "exp_array.txt",
perm = 1000,
QN = T)
dim(results)
# [1] 104 25
results[1:4,1:4]
# B cells naive B cells memory Plasma cells T cells CD8
# SUB137169 0.04924204 0 0.0000000000 0.2643171
# SUB137172 0.07936758 0 0.0147950385 0.2623424
# SUB137208 0.03038180 0 0.0004003184 0.1864818
# SUB137209 0.07007071 0 0.0089139936 0.1127511
|
更新:immunedeconv包#
集成了多种常见的免疫浸润分析算法,提供了便捷的分析接口
1
2
3
4
5
6
7
|
# 安装方式
## (1) 官方推荐在linux使用conda安装
conda install -c bioconda -c conda-forge r-immunedeconv
## (2) 经实操后,window上也可正常使用
install.packages("remotes")
remotes::install_github("omnideconv/immunedeconv")
|
1、内置算法类型#
1.1 按支持物种分#
1
2
3
4
5
6
7
8
9
10
11
|
deconvolution_methods
# quantiseq
# timer
# cibersort
# cibersort_abs
# mcp_counter
# xcell
# epic
# abis
# consensus_tme
# estimate
|
1
2
3
4
5
|
deconvolution_methods_mouse
# mmcp_counter
# seqimmucc
# dcq
# base
|
1.2 按结果解读分#
1
2
3
4
5
6
7
8
|
# MCP-counter
# xCell
# TIMER
# ConsensusTME
# ESTIMATE
# ABIS
# mMCP-counter (mouse based)
# BASE (mouse based)
|
1
2
|
# CIBERSORT
# DCQ (mouse based)
|
1
2
3
4
|
# EPIC
# quanTIseq
# CIBERSORT abs. mode
# seqImmuCC (mouse based)
|
1.3 算法细胞类型#
immunedeconv包一方面可以将每种算法的原始结果作为输出,另一方面也综合所有算法的细胞类型进行了细胞类型统一。
1
2
3
4
5
6
7
8
|
cell_types = readxl::read_xlsx("cell_type_mapping.xlsx",sheet = "mapping")
## (1) 原始细胞类型
cell_types %>%
dplyr::distinct(method_dataset, method_cell_type)
## (2) 统一细胞类型
cell_types %>%
dplyr::distinct(method_dataset, cell_type)
|
2、基本使用方式#
Step1:准备基因表达矩阵
- 行名是基因(symbol)、列名是样本名的表达矩阵;
- 一般情况下需要进行TPM标准化,但不需进一步log处理
- 对于xcell、MCP-counter算法不进行标准化也可
1
2
3
4
5
6
7
8
|
# 包示例数据
expr_mat = dataset_racle$expr_mat
head(expr_mat)
# LAU125 LAU355 LAU1255 LAU1314
# A1BG 0.82 0.58 0.81 0.71
# A1CF 0.00 0.01 0.00 0.00
# A2M 247.15 24.88 2307.94 20.30
# A2M-AS1 1.38 0.20 2.60 0.28
|
Step2:选择合适算法,直接运行即可得到结果
每一种算法均有两种使用方式,以xcell算法为例
- (1)
deconvolute(),其分析结果为统一后的细胞类型
1
2
3
4
5
6
|
method = "xcell"
scores = deconvolute(expr_mat, method=method)
dim(scores)
# [1] 39 5
## deconvolute_mouse()
|
- (2)
deconvolute_xcell()函数,其分析结果是原始结果
1
2
3
|
scores = deconvolute_xcell(expr_mat, arrays=F)
dim(scores)
# [1] 67 4
|
目前感觉更推荐第二种使用方式。(1)首先输出结果为原始算法类型,(2)函数的参数为针对该算法所设
3、特殊使用方式#
(1)对于一些算法,表达数据类型如果是芯片数据需要特别声明;例如quantiseq、cibersort、xcell等
1
2
|
method = "quantiseq"
scores = deconvolute(expr_mat, method = method, arrays = FALSE)
|
(2)对于timer算法,需要交代每个样本的样本类型
1
2
3
4
5
6
|
method = "timer"
method = "ConsensusTME"
# 所支持的样本类型
# https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations
timer_available_cancers
scores = deconvolute(expr_mat, method = method, indications = c("kich", "blca", "brca", "brca"))
|
(3)对于cibersort算法,需要提供两个文件的路径
1
2
3
4
5
|
method = "cibersort"
method = "cibersort_abs"
set_cibersort_binary("./CIBERSORT1.04.R")
set_cibersort_mat("./LM22.txt")
scores = deconvolute(expr_mat, method = method)
|
(4)ESTIMATE算法用于预测肿瘤样本的肿瘤、免疫、基质分数以及肿瘤纯净度
1
2
3
4
5
6
|
scores = deconvolute_estimate(expr_mat)
# LAU125 LAU355 LAU1255 LAU1314
# StromalScore -1294.7848659 -653.7842997 -499.1445971 -1144.8145113
# ImmuneScore 17.3822270 3722.9742037 3033.2695535 3550.9927343
# ESTIMATEScore -1277.4026389 3069.1899040 2534.1249564 2406.1782230
# TumorPurity 0.9141139 0.4927852 0.5595386 0.5750048
|
对于小鼠表达数据,可以使用专门针对鼠的算法,也可将其基因名进行同源转换,再使用针对人的算法。在针对鼠的算法中, seqimmucc支持两种不同方式(“SVR”/“LLSR”)。对于前者,需要按照CIBERSORT流程声明两个文件的路径
(5)其中有4种算法支持用户提供自定义的细胞类型特征进行预测
- base: deconvolute_base_custom()
- cibersort norm/abs: deconvolute_cibersort_custom()
- epic: deconvolute_epic_custom()
- consensus_tme: deconvolute_consensus_tme_custom()