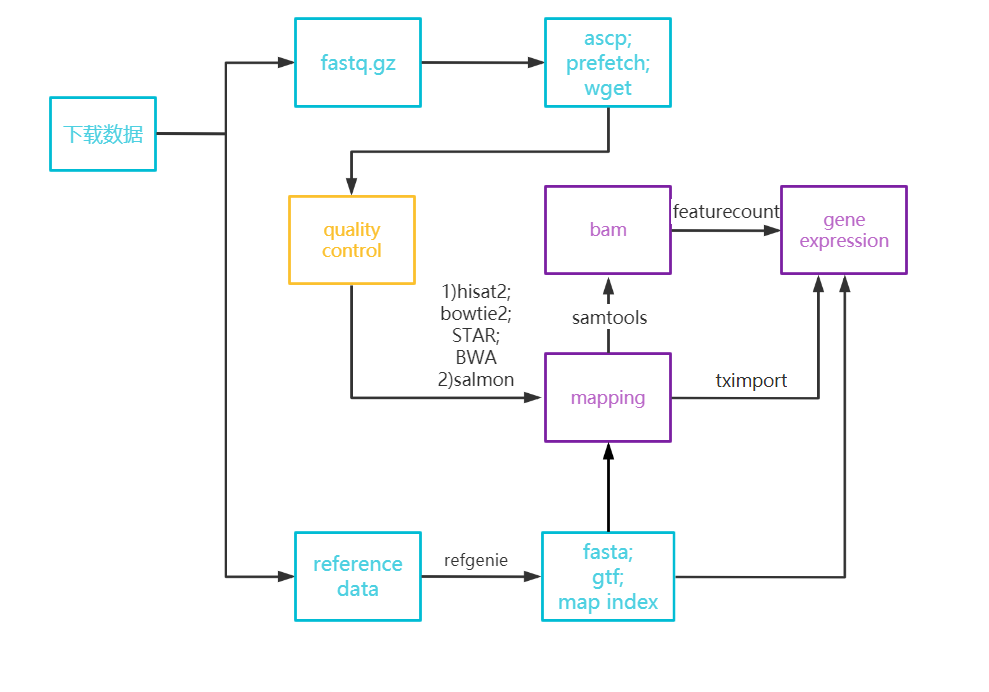
RNA-seq数据比对流程主要分为三步(1)整理数据;(2)质控;(3)比对。其中每一步都涉及到若干软件的用法,如下简单整理基本的分析流程。
示例数据:GSE158623中6个样本的RNA-seq测序结果(human),对应SRR12720999~SRR12721004
1
2
3
4
5
6
7
|
cat SraAccList.txt
SRR12720999
SRR12721000
SRR12721001
SRR12721002
SRR12721003
SRR12721004
|
0、搭建conda环境#
根据需要,搭建两个conda分析环境
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
|
### (1) 下载数据
conda activate download
#ascp的aspera高速下载
conda install -c hcc aspera-cli
#prefech下载sra文件
conda install -c bioconda sra-tools
# 基因组下载商店
# conda install refgenie
# QC质控
conda install -c bioconda trim-galore multiqc
### (2) 比对分析
conda activate fq_map
#不同类型的比对软件
conda install -c bioconda hisat2
conda install -c bioconda star=2.7.1a
conda install -c bioconda bwa
conda install -c bioconda bowtie2
conda install -c bioconda salmon=1.5.2
# 汇总比对结果
conda install -c bioconda multiqc
# 基因组下载商店
# conda install refgenie
# sam2bam转换
conda install -c bioconda samtools
# 基因表达定量
conda install -c bioconda subread
|
如上,默认下载软件的最新版本即可,但结合尝试、探索,star与salmon比对软件的版本信息必须与构建对应索引文件的软件版本一致(refgenie构建)。因此安装了上述指定的版本。
1、下载数据#
(1)下载公共测序数据:参考之前笔记,有aspera/prefetch/wget等三种下载方式,如下为aspera的下载代码
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
# 批量生成下载链接
# era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR166/009/SRR1663609/SRR1663609_1.fastq.gz
touch ascp.link
cat SraAccList.txt | while read id
do
echo "era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/${id:0:6}/0${id:0-2}/${id}/${id}_1.fastq.gz" >> ascp.link
echo "era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/${id:0:6}/0${id:0-2}/${id}/${id}_2.fastq.gz" >> ascp.link
done
#ascp高速下载
cat ascp.link |while read sample
do
ascp -QT -l 300m -P33001 \
-i ~/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh \
$sample .
done
|
(2)下载基因组相关参考数据:主要包括基因组序列fasta、基因组注释文件gtf、各类比对软件的基因组索引文件。
参考之前笔记,上述文件均可通过refgenie工具下载。如下为下载代码
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
refgenie init -c ~/refgenie/genome_config.yaml
refgenie listr -c ~/refgenie/genome_config.yaml
refgenie listr -g hg38 -c ~/refgenie/genome_config.yaml
#参考基因组
refgenie pull hg38/fasta -c ~/refgenie/genome_config.yaml
refgenie pull hg38_cdna/fasta -c ~/refgenie/genome_config.yaml
#参考注释信息
refgenie pull hg38/gencode_gtf -c ~/refgenie/genome_config.yaml
#比对软件的索引文件
refgenie pull hg38/bowtie2_index -c ~/refgenie/genome_config.yaml
refgenie pull hg38/bwa_index -c ~/refgenie/genome_config.yaml
refgenie pull hg38/star_index -c ~/refgenie/genome_config.yaml
refgenie pull hg38/hisat2_index -c ~/refgenie/genome_config.yaml
refgenie pull hg38_cdna/salmon_index -c ~/refgenie/genome_config.yaml
#列出本地已经下载的数据
refgenie list -c ~/refgenie/genome_config.yaml
|
2、测序数据质控#
主要目的是为了过滤测序数据中的低质量reads。
相关RNAseq质控软件有很多,如下以trim_glaore软件用法为例。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
# (1)以其中一个作为示例
pare_dir=/home/data/****/mapping
fq1=${pare_dir}/raw/ebi/SRR12720999_1.fastq.gz
fq2=${pare_dir}/raw/ebi/SRR12720999_2.fastq.gz
trim_galore -j 8 -q 25 --phred33 --length 36 \
-paired -o ${pare_dir}/trim \
$fq1 $fq2
# (2)批量分析
cat ${pare_dir}/SraAccList.txt | while read id
do
echo $id
trim_galore -j 8 -q 25 --phred33 --length 36 \
-paired -o ${pare_dir}/trim \
${pare_dir}/raw/ebi/${id}_1.fastq.gz \
${pare_dir}/raw/ebi/${id}_2.fastq.gz
done
# (3)查看质控之后的fatsq.gz质量
fastqc $(ls ${pare_dir}/trim/*gz) -o ${pare_dir}/trim/ -t 10
multiqc ./ -n trim_multiqc_report.html
|
3、各类比对软件#
- RNAseq比对软件有很多,常见的包括hisat2、star、bowtie2、bwa以及salmon;
- 命令调用均主要包括参数:参考基因组索引文件、 测序数据、输出结果名,以及线程数
- 比对结果通常是
.sam文件,一般需要转为.bam格式,最后提取出count表达数据;
(1)以其中一对fatsq.gz文件为例,总结各个比对软件的用法

1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
|
conda activate fq_map
pare_dir=/home/data/****/mapping
fq1=${pare_dir}/trim/SRR12720999_1_val_1.fq.gz
fq2=${pare_dir}/trim/SRR12720999_2_val_2.fq.gz
#hisat2
ref_idx_hisat2=$(refgenie seek hg38/hisat2_index -c ~/refgenie/genome_config.yaml)
time hisat2 -t -p 10 -x $ref_idx_hisat2 -1 $fq1 -2 $fq2 -S test.sam
#STAR
ref_idx_star=$(refgenie seek hg38/star_index -c ~/refgenie/genome_config.yaml)
time STAR --genomeDir $ref_idx_star \
--runThreadN 10 --readFilesCommand zcat \
--readFilesIn $fq1 $fq2 \
--outSAMtype SAM --outFileNamePrefix test
#Bowtie2
ref_idx_bowtie2=$(refgenie seek hg38/bowtie2_index -c ~/refgenie/genome_config.yaml)
time bowtie2 -p 10 -x $ref_idx_bowtie2 -1 $fq1 -2 $fq2 -S test.sam
#BWA
ref_idx_bwa=$(refgenie seek hg38/bwa_index -c ~/refgenie/genome_config.yaml)
time bwa mem -t 10 $ref_idx_bwa $fq1 $fq2 -o test.sam
#salmon
ref_idx_salmon=$(refgenie seek hg38_cdna/salmon_index -c ~/refgenie/genome_config.yaml)
time salmon quant -i $ref_idx_salmon -l A \
-1 $fq1 -2 $fq2 \
-p 10 -o test_quant
|
(2)基于全部数据,总结每个比对软件的批量比对全流程用法
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
|
#1、定义变量
ref_idx_hisat2=$(refgenie seek hg38/hisat2_index -c ~/refgenie/genome_config.yaml)
ref_gtf=$(refgenie seek hg38/gencode_gtf -c ~/refgenie/genome_config.yaml)
pare_dir=/home/data/****/mapping
#2、批量比对
cat ${pare_dir}/SraAccList.txt | while read id
do
echo $id
#首先进行比对,生成sam文件
echo "Start hisat mapping...."
hisat2 -p 10 -x $ref_idx_hisat2 \
-1 ${pare_dir}/trim/${id}_1_val_1.fq.gz \
-2 ${pare_dir}/trim/${id}_2_val_2.fq.gz \
-S ${pare_dir}/hisat2/${id}.sam
#然后sam转bam,同时删除内存较大的bam
echo "Start sam2bam...."
samtools view -S ${pare_dir}/hisat2/${id}.sam \
-@ 10 -b > ${pare_dir}/hisat2/${id}.bam
rm ${pare_dir}/hisat2/${id}.sam
done
#3、featureCounts提取表达信息
featureCounts -p -T 10 -t exon -g gene_name -a $ref_gtf -o hisat2_exp_counts.txt *.bam
#第1列为基因名,第7至最后1列为各个样本的表达信息
#4、各个样本比对率概况
multiqc ./ -n hisat2_multiqc_report.html
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
|
#1、定义变量
ref_idx_star=$(refgenie seek hg38/star_index -c ~/refgenie/genome_config.yaml)
ref_gtf=$(refgenie seek hg38/gencode_gtf -c ~/refgenie/genome_config.yaml)
pare_dir=/home/data/****/mapping
#2、批量比对
# 尝试换用for循环,本质都一样
for sample in $(cat ${pare_dir}/SraAccList.txt)
do
echo "START sample ${sample}"
echo "Start STAR mapping..."
#首先进行比对,生成sam文件
STAR --genomeDir $ref_idx_star \
--runThreadN 10 --readFilesCommand zcat \
--readFilesIn ${pare_dir}/trim/${sample}_1_val_1.fq.gz ${pare_dir}/trim/${sample}_2_val_2.fq.gz \
--outSAMtype SAM --outFileNamePrefix ${sample}
echo "Start sam2bam..."
#然后sam转bam,同时删除内存较大的bam
samtools view -S ${pare_dir}/star/${sample}Aligned.out.sam \
-@ 10 -b > ${pare_dir}/star/${sample}.bam
rm ${pare_dir}/star/${sample}Aligned.out.sam
done
#3、featureCounts提取表达信息
featureCounts -p -T 10 -t exon -g gene_name -a $ref_gtf -o star_exp_counts.txt *.bam
#第1列为基因名,第7至最后1列为各个样本的表达信息
#4、各个样本比对率概况
multiqc ./ -n star_multiqc_report.html
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
|
#1、定义变量
ref_idx_bowtie2=$(refgenie seek hg38/bowtie2_index -c ~/refgenie/genome_config.yaml)
ref_gtf=$(refgenie seek hg38/gencode_gtf -c ~/refgenie/genome_config.yaml)
pare_dir=/home/data/****/mapping
#2、批量比对
for sample in $(cat ${pare_dir}/SraAccList.txt)
do
echo "START sample ${sample}"
echo "Start Bowtie2 mapping..."
bowtie2 -p 10 -x $ref_idx_bowtie2 \
-1 ${pare_dir}/trim/${sample}_1_val_1.fq.gz \
-2 ${pare_dir}/trim/${sample}_2_val_2.fq.gz \
-S ${pare_dir}/bowtie2/${sample}.sam
echo "Start sam2bam..."
samtools view -S ${pare_dir}/bowtie2/${sample}.sam \
-@ 10 -b > ${pare_dir}/bowtie2/${sample}.bam
rm ${pare_dir}/bowtie2/${sample}.sam
done
#3、featureCounts提取表达信息
featureCounts -p -T 10 -t exon -g gene_name -a $ref_gtf -o bowtie2_exp_counts.txt *.bam
#第1列为基因名,第7至最后1列为各个样本的表达信息
#4、各个样本比对率概况
multiqc ./ -n bowtie2_multiqc_report.html
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
|
#1、定义变量
ref_idx_bwa=$(refgenie seek hg38/bwa_index -c ~/refgenie/genome_config.yaml)
ref_gtf=$(refgenie seek hg38/gencode_gtf -c ~/refgenie/genome_config.yaml)
pare_dir=/home/data/****/mapping
#2、批量比对
for sample in $(cat ${pare_dir}/SraAccList.txt)
do
echo "START sample ${sample}"
echo "Start BWA mapping..."
bwa mem -t 10 $ref_idx_bwa \
${pare_dir}/trim/${sample}_1_val_1.fq.gz \
${pare_dir}/trim/${sample}_2_val_2.fq.gz \
-S -o ${pare_dir}/bwa/${sample}.sam
echo "Start sam2bam..."
samtools view -S ${pare_dir}/bwa/${sample}.sam \
-@ 10 -b > ${pare_dir}/bwa/${sample}.bam
rm ${pare_dir}/bwa/${sample}.sam
done
#3、featureCounts提取表达信息
featureCounts -p -T 10 -t exon -g gene_name -a $ref_gtf -o bwa_exp_counts.txt *.bam
#第1列为基因名,第7至最后1列为各个样本的表达信息
#4、各个样本比对率概况
multiqc ./ -n bwa_multiqc_report.html
|
1
2
3
4
5
6
7
8
9
10
11
12
13
|
#1、定义变量
refgenie list -g hg38_cdna -c ~/refgenie/genome_config.yaml
ref_idx_salmon=$(refgenie seek hg38_cdna/salmon_index -c ~/refgenie/genome_config.yaml)
pare_dir=/home/data/****/mapping
#2、批量比对
cat ${pare_dir}/SraAccList.txt | while read id
do
echo $id
salmon quant -i $ref_idx_salmon -l A \
-1 ${pare_dir}/trim/${id}_1_val_1.fq.gz \
-2 ${pare_dir}/trim/${id}_2_val_2.fq.gz \
-p 10 -o ${pare_dir}/salmon/${id}_quant
done
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
|
library(tximport)
library(readr)
library(biomaRt)
ensembl = useMart("ensembl",dataset="hsapiens_gene_ensembl")
attributes = listAttributes(ensembl)
attributes[1:5,]
# library(httr)
# httr::set_config(config(ssl_verifypeer = 0L))
gene_ids <- getBM(attributes= c("hgnc_symbol","ensembl_transcript_id"),
mart= ensembl)
gene_ids = gene_ids[!duplicated(gene_ids[,2]),]
colnames(gene_ids) = c("gene_id","tx_id")
gene_ids = gene_ids[,c("tx_id","gene_id")]
files <- list.files(pattern = '*sf',recursive = T, full.names=T)
txi <- tximport(files, type = "salmon", tx2gene = gene_ids, ignoreTxVersion = T, ignoreAfterBar=T)
class(txi)
names(txi)
head(txi$length)
head(txi$counts)
srrs = stringr::str_extract(files, "SRR[:digit:]+")
salmon_expr <- txi$counts
salmon_expr <- apply(salmon_expr, 2, as.integer)
rownames(salmon_expr) <- rownames(txi$counts)
colnames(salmon_expr) <- srrs
save(salmon_expr, file="./salmon_expr.rda")
|