1、背景知识
(1)两种富集分析
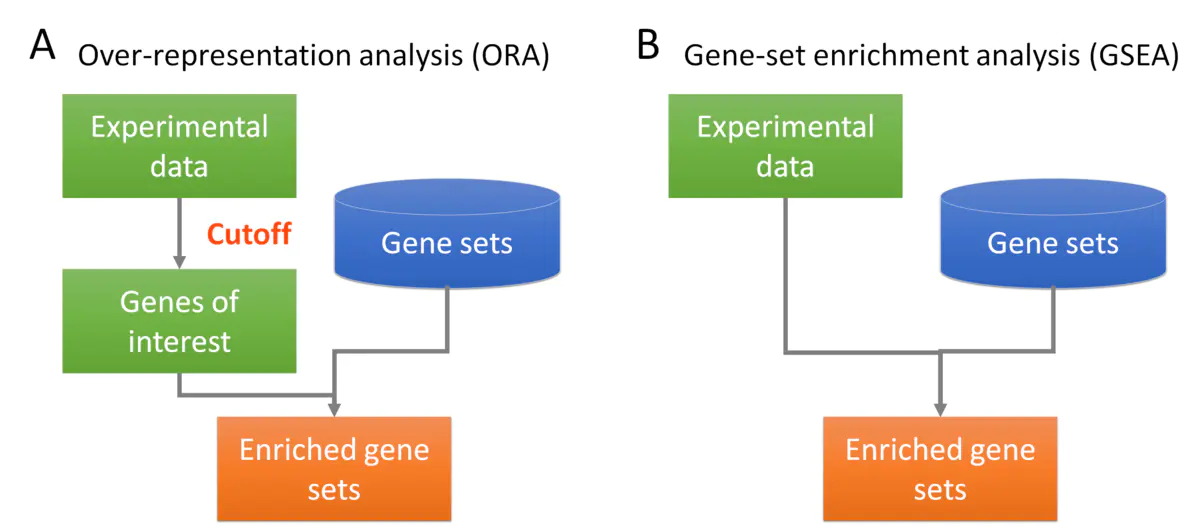
- 基于超几何检验的ORA(over representation analysis)富集分析
① 假设对转录组分组测序的10000个基因表达数据进行差异分析;
② 按照规定cutoff阈值(logFC/Pvalue)筛选得到100个差异表达基因;
③ 对于特定通路,包含500个基因;其中有30个属于差异基因

④ 差异基因是否富集到该通路,即表示30/70与470/9430相比,是否具有显著意义。
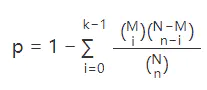
|
|
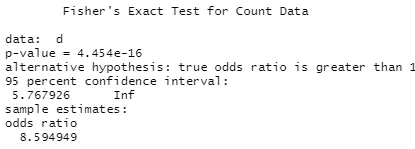
如上计算结果,p值越小(<0.05),说明得到的差异基因是显著富集到这个通路基因集上的。然后就可以这个基因集的生物学意义讲述后续的故事了。
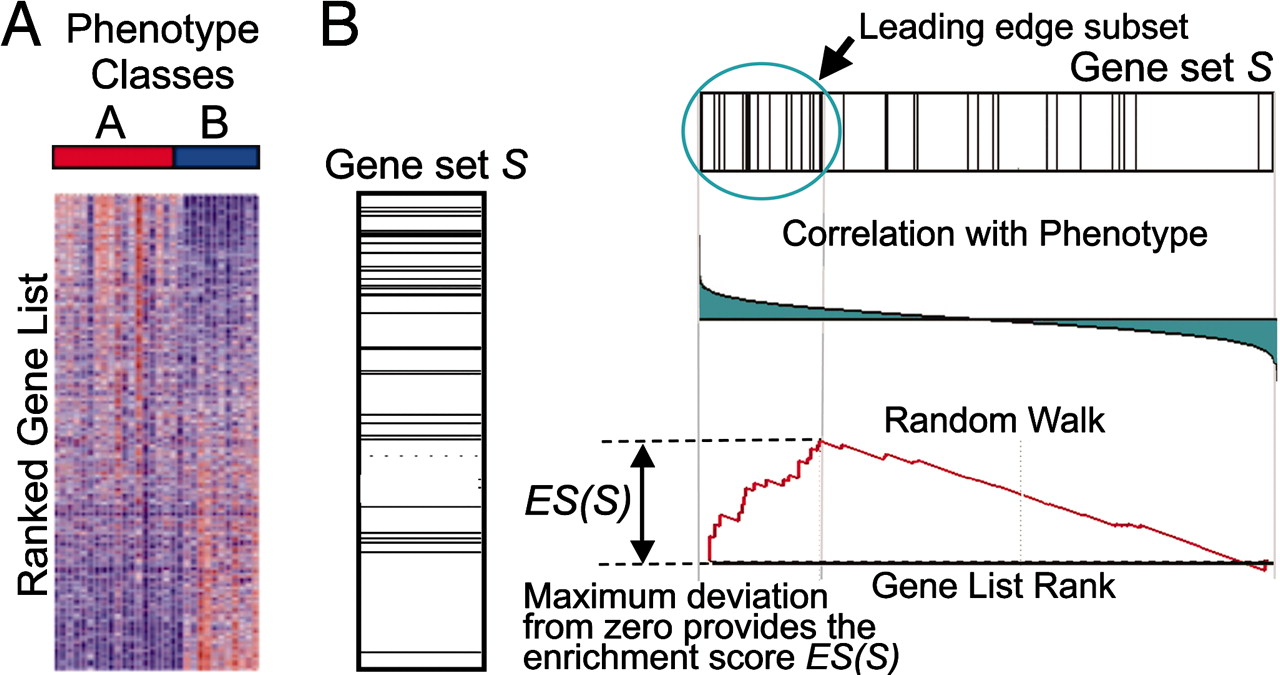
- 基于置换检验的GSEA(gene set enrichment analysis)富集分析
① 假设对转录组分组测序的10000个基因表达数据进行差异分析,一般按照差异倍数从高到低进行排序;
② 假设对于包含500个基因的特定通路,遍历上述10000个基因的排序列表;
③ 在遍历过程中,如基因属于通路集则加分(hit),不属于则减分;加减分的权重与差异倍数先关。
④ ES(enrichment score)表示遍历过程中产生的绝对值最大值。ES大于0,表明通路集富集到排序列表的顶部;反之则富集到排序列表的底部。
⑤ 随机选择500个基因若干次,计算ES背景分布,从而计算相应的P值。
(2)通路基因集
常见的通路基因集包括GO、KEGG、Reactome等。MsigDB数据库则综合收集了多种通路基因集。4
-
GO
- Gene Ontology 基因本体论
- http://www.geneontology.org/
- GO 分别定义了三种类型的基因集:①细胞组成(cellular component,CC);②生物过程(biological process,BP);③分子功能(Molecular Function,MF)。详见http://geneontology.org/docs/ontology-documentation/
- 举个例子来说:基因A(其蛋白产物是cytochrome c(细胞色素c)),该基因的具有氧化还原活性(molecular function);参与氧化磷酸化生物学过程(molecular function);而发挥作用的位置位于细胞线粒体基质(cellular component)
-
KEGG
- Kyoto Encyclopedia of Genes and Genomes 京都基因与基因组百科全书
- https://www.genome.jp/kegg/
- KEGG是系统分析基因功能,联系基因组信息和功能信息,以探索分子相互作用和反应网络的知识库(人工绘制),其中包含有大量的通路(PATHWAY)图,涉及metabolism, genetic and environmental information processing, cellular processes, organismal systems, human diseases, and drug development七大类。
-
Reactome https://reactome.org/
-
WikiPathway https://www.wikipathways.org/
-
Msigdb
- Molecular Signatures Database 分子特征数据库
- https://www.gsea-msigdb.org/gsea/msigdb/index.jsp
- 截止目前该数据库收集有32284个基因集,可分为9大类 — H: hallmark gene sets,C1: positional gene sets,C2: curated gene sets,C3: regulatory target gene sets,C4: computational gene sets,C5: ontology gene sets,C6: oncogenic signature gene sets,C7: immunologic signature gene sets,C8: cell type signature gene sets
此外还有其它类型通路基因集,例如疾病基因集DO,就不一一列举了。
(3)clusterProfiler包
R包clusterProfiler由南方医科大学余光创团队开发,是一个常用的富集分析工具包,目前已更新到4.x系列版本。该包包含了富集分析相关的方方面面函数功能,从上游分析到下游可视化等一应俱全。后续笔记主要参考官方教程学习该包的基础用法。
- DOI: 10.1016/j.xinn.2021.100141
- Tutorial:https://yulab-smu.top/biomedical-knowledge-mining-book/index.html
2、富集分析
|
|
需要使用ENTREZID的基因ID。如果是其它格式,可使用
clusterProfiler::bitr()函数或者之前笔记提到的方式进行转换。
1 2 3 4 5 6 7 8 9 10keytypes(org.Hs.eg.db) # [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" # [6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME" # [11] "GENETYPE" "GO" "GOALL" "IPI" "MAP" # [16] "OMIM" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" # [21] "PMID" "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" # [26] "UNIPROT" ids <- bitr(x, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
(1)GO
- ORA
|
|
- GO 去冗余
|
|
- GSEA
|
|
(2)KEGG
- ORA
|
|
当出现报错类似于No gene can be mapped when using enrichKEGG,且检查输入基因无误后,一个可能的解决方案是自己构建本地KEGG数据库。参考https://github.com/YuLab-SMU/clusterProfiler/issues/561中undo6411解答。或者也可以直接从MsigDB数据库下载,再使用
GSEA()函数。
- GSEA
|
|
针对其它通路集的富集分析函数
通路集 ORA函数 GSEA函数 WikiPathway enrichWP gseWP Reactome enrichPathway gsePathway DO(disease ontology) enrichDO gseDO NCG(Network of Cancer Gene) enrichNCG gseNCG DisGeNET(disease gene network) enrichDGN gseDGN Mesh(医学主题词) enrichMeSH gseMeSH
(3)自定义基因集
- clusterProfiler包提供的
enricher()与GSEA()函数可实现对自定义基因集进行富集分析 - 主要通过TERM2GENE参数提供基因集数据框,一列名为term代表通路名;一列为gene代表组成基因
- 如下以MsigDB数据库为例,学习用法。该库的详细介绍参见笔记
|
|
- 富集分析
|
|
3、可视化
- 以上面分析的ego与gseago为例
|
|
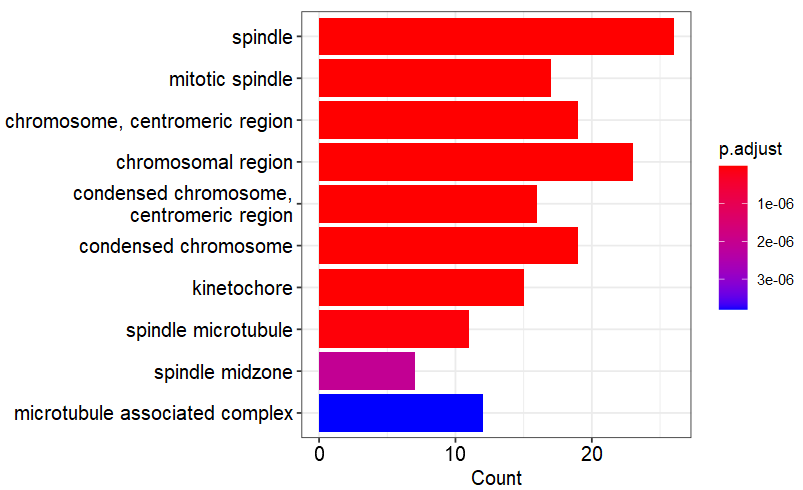
(1)柱状图/点图
|
|
|
|
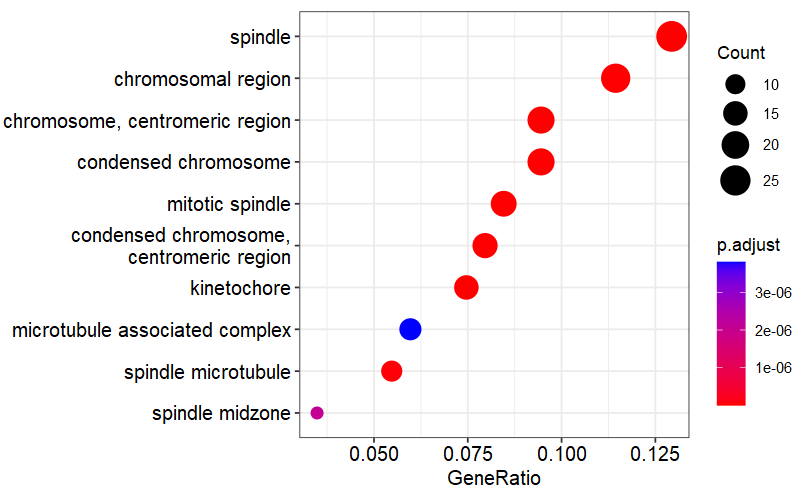
|
|
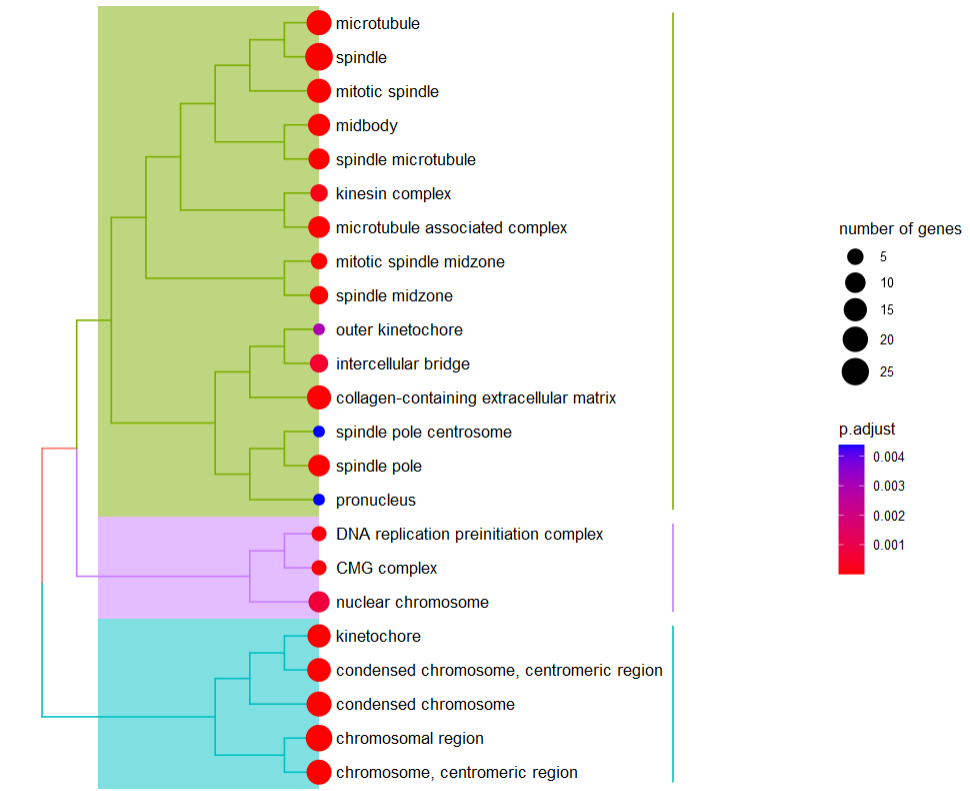
(2)GSEA打分
- 单个通路GSEA
|
|
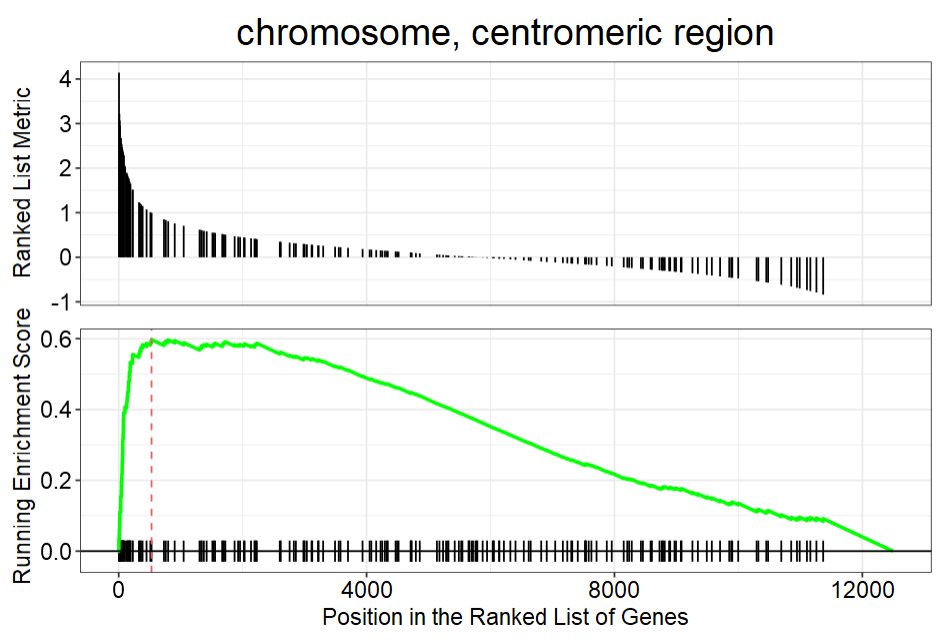
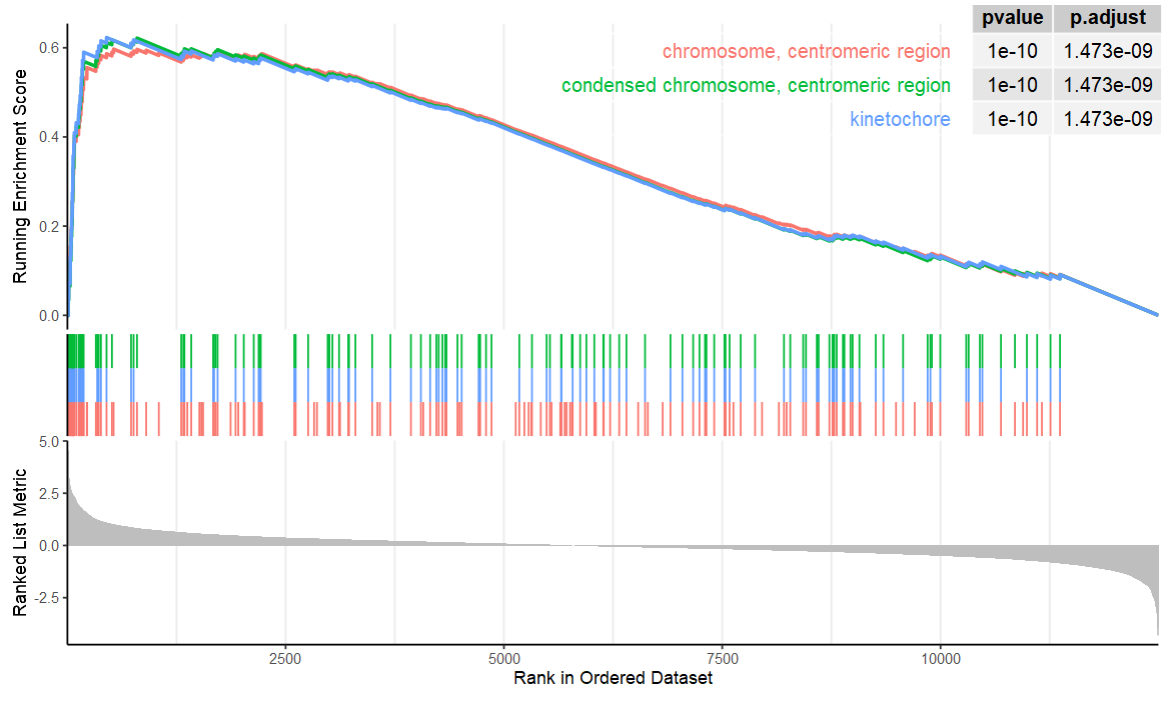
- 多条通路结果
|
|
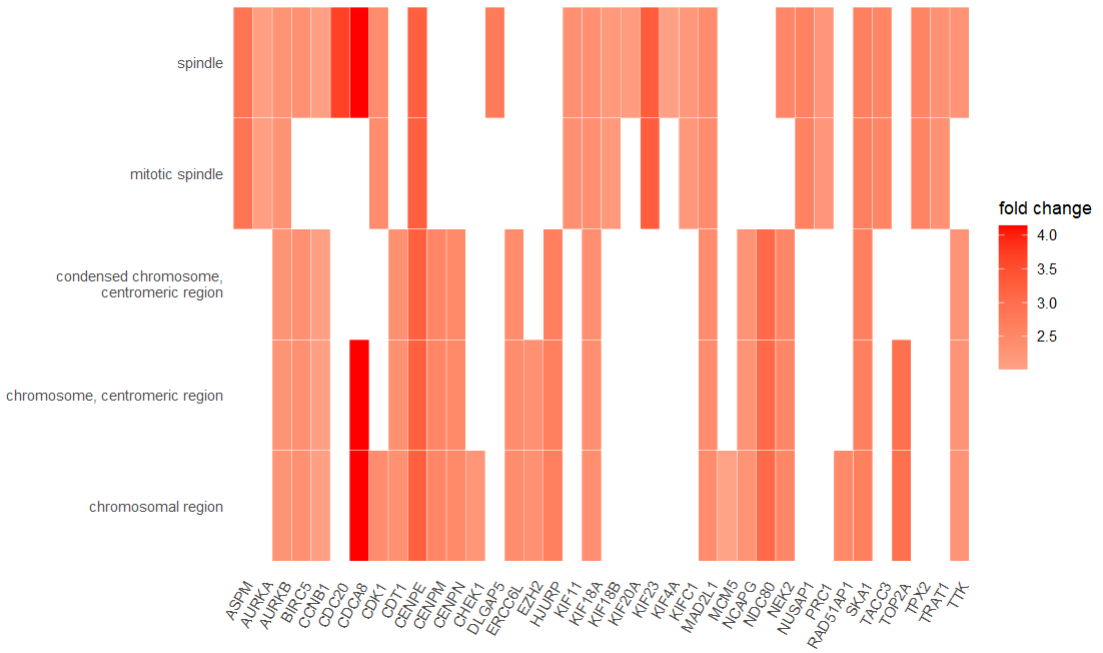
(3)通路与基因
- 热图
|
|
- 网络图
|
|
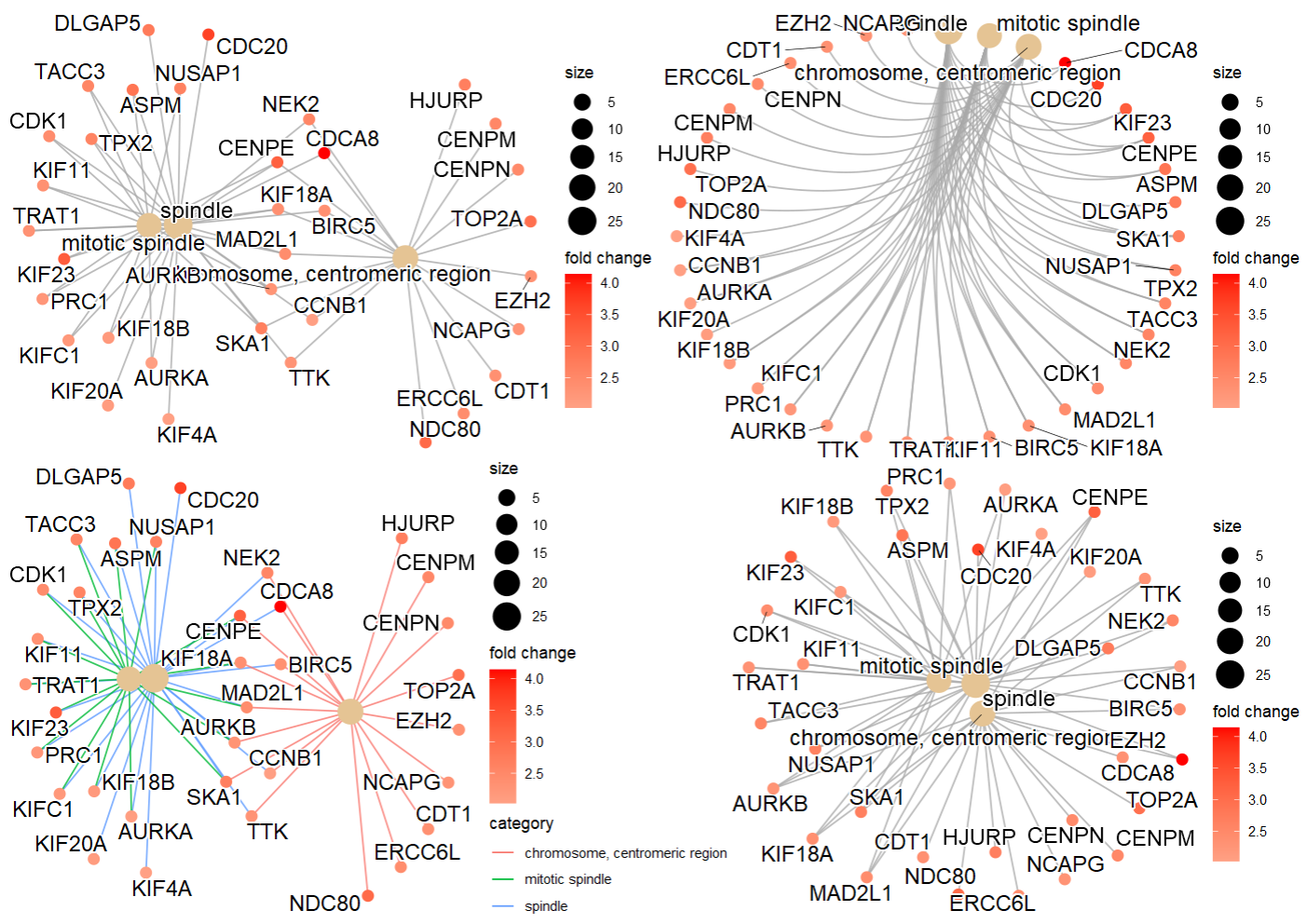
(4)通路与通路
|
|
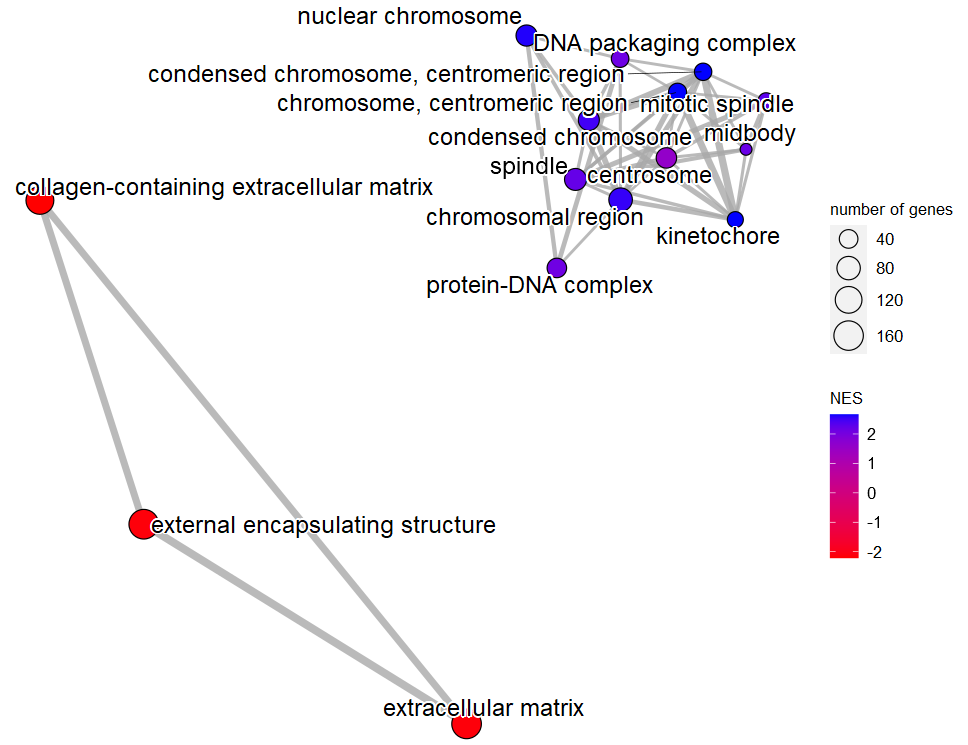
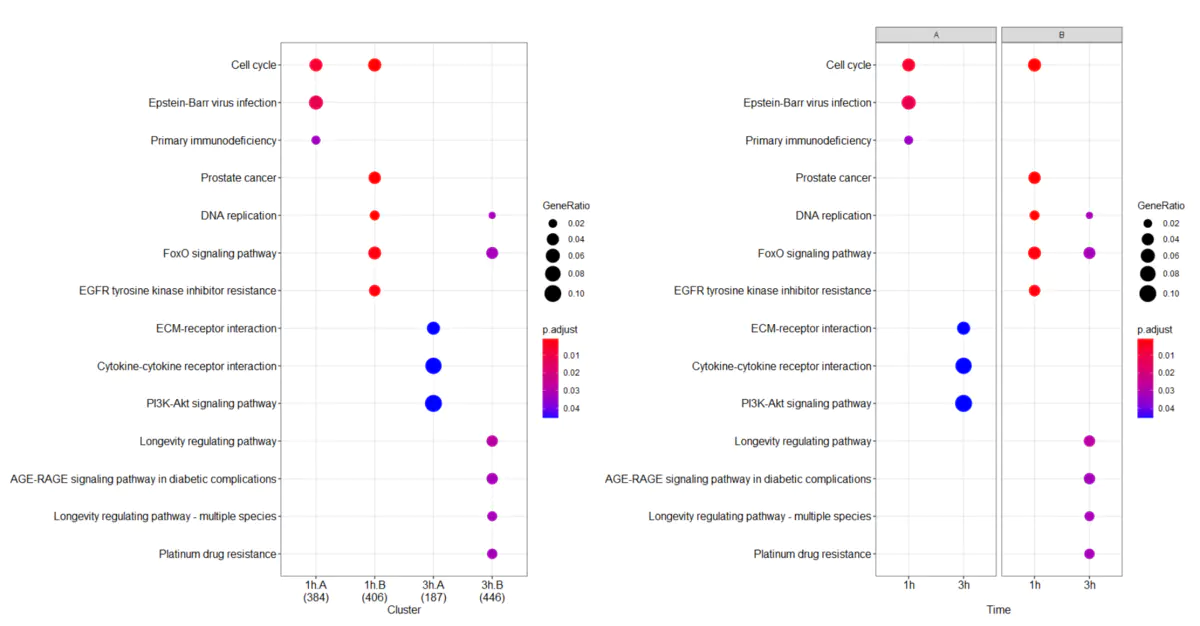
4、多基因集分析
- clusterProfiler包的
compareCluster()函数提供了针对多组实验设计得到的若干差异基因列表的同时富集分析(ORA)结果,及友好的可视化。
|
|
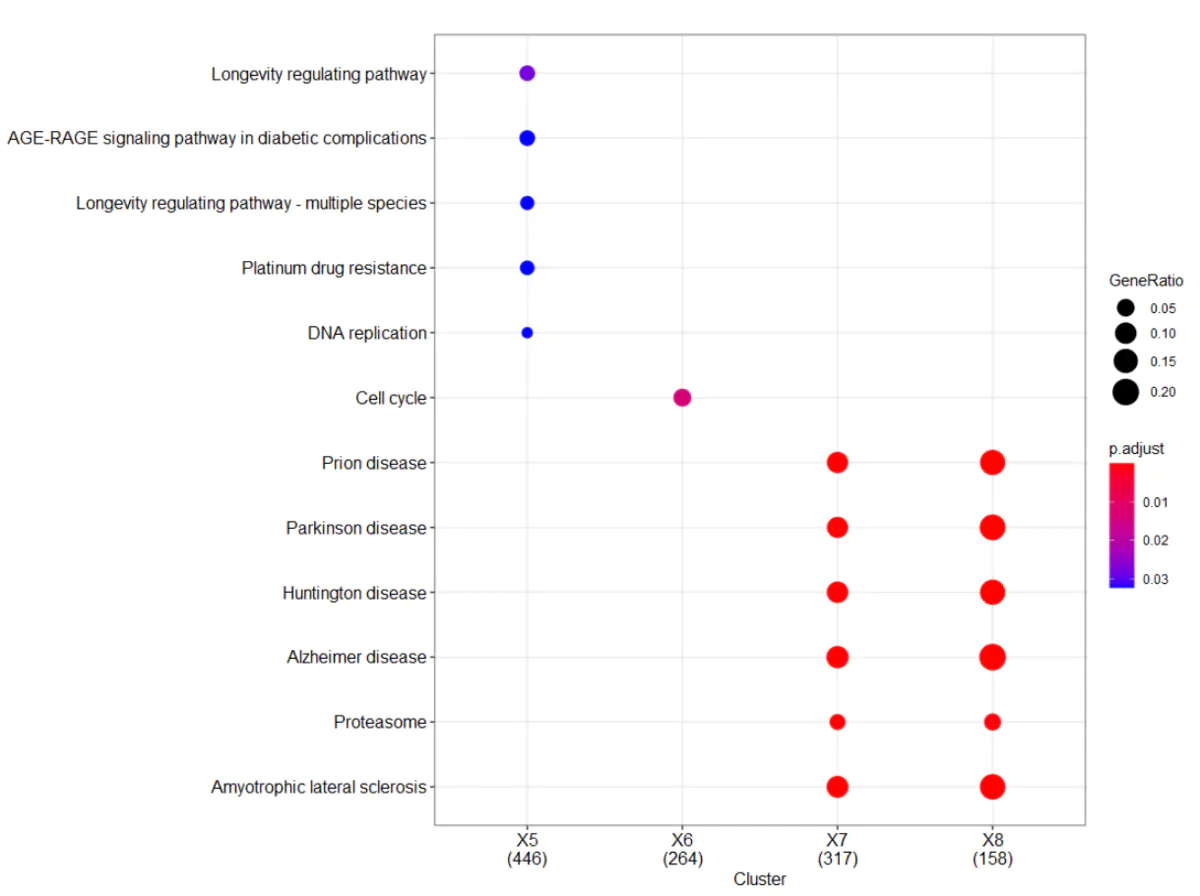
|
|