以前通路富集分析直接使用clusterprofiler包,阅读文献发现GSEA分析及可视化较多使用Broad团队研发的工具,现简要学习其(window版本)使用方法。
软件及示例数据下载:https://www.gsea-msigdb.org/gsea/index.jsp
初始论文:https://www.pnas.org/content/102/43/15545 2005
官方手册:https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html
视频教程:https://www.youtube.com/watch?v=KY6SS4vRchY (推荐)

1、数据准备#
GSEA分析需要两大类数据:两组样本的表达矩阵,通路基因集
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
|
# BiocManager::install("airway")
library(tidyverse)
library(airway)
data(airway)
exp=assay(airway)
class(exp)
# [1] "matrix" "array"
dim(exp)
# [1] 64102 8
exp[1:4,1:4]
## ID转换
library(org.Hs.eg.db)
gene_ids = AnnotationDbi::select(org.Hs.eg.db, keys=rownames(exp),
columns=c("SYMBOL"),
keytype="ENSEMBL")
gene_ids = gene_ids %>%
dplyr::distinct(ENSEMBL, .keep_all = T) %>%
dplyr::distinct(SYMBOL, .keep_all = T) %>%
na.omit()
exp = exp[match(gene_ids$ENSEMBL, rownames(exp)),]
exp[1:4,1:4]
rownames(exp) = gene_ids$SYMBOL
exp[1:4,1:4]
## 分组信息
meta = colData(airway)[,3,drop=F] %>%
as.data.frame() %>%
dplyr::arrange(dex)
colnames(meta) = "Group"
exp = exp[, rownames(meta)]
identical(colnames(exp), rownames(meta))
|
(1)表达矩阵导出为gct格式#

1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = exp,
colData = meta,
design=~Group)
dds <- DESeq(dds)
norm_counts <- counts(dds, normalized = T) %>%
as.data.frame() %>%
tibble::rownames_to_column("NAME") %>%
dplyr::mutate(description=NAME, .before=2)
norm_counts[1:4,1:4]
# NAME description SRR1039509 SRR1039513
# 1 TSPAN6 TSPAN6 500.2045 610.3226
# 2 TNMD TNMD 0.0000 0.0000
# 3 DPM1 DPM1 575.0119 545.9994
# 4 SCYL3 SCYL3 235.5874 245.3257
fid <- "norm_counts.gct"
writeLines(c("#1.2", paste(nrow(norm_counts), ncol(norm_counts) - 2, collapse="\t")),
fid, sep="\n")
write.table(norm_counts, fid, sep = "\t",
quote = F, row.names = F,
append = TRUE)
|
(2)分组信息导出为cls格式#

1
2
3
4
5
|
fid <- "norm_counts.cls"
writeLines(c(paste(nrow(meta), length(unique(meta$Group)), 1),
paste("#", unique(meta$Group)[1], unique(meta$Group)[2]),
paste(as.character(meta$Group), collapse = " ")),
fid, sep="\n")
|
(3)通路集gmt格式#
- GSEA软件提供了msigdb的8类通路基因集(symbol ID的gmt格式),可直接使用
- 如需提供自定义的基因集,也需要储存为gmt格式
1
2
3
4
5
6
7
8
9
10
11
12
13
|
pw_gene = clusterProfiler::read.gmt("h.all.v7.5.1.symbols.gmt")
pw_gene.list = split(pw_gene$gene, pw_gene$term)
lapply(seq(pw_gene.list), function(i){
# i = 1
pw_name = names(pw_gene.list)[i]
pw_gene = pw_gene.list[[i]]
pw_sle = paste(pw_name,pw_name,
paste(pw_gene, collapse = " "),
collapse = " ")
return(pw_sle)
}) %>% unlist %>%
writeLines(., "test.gmt", sep="\n")
|
2、分析步骤#
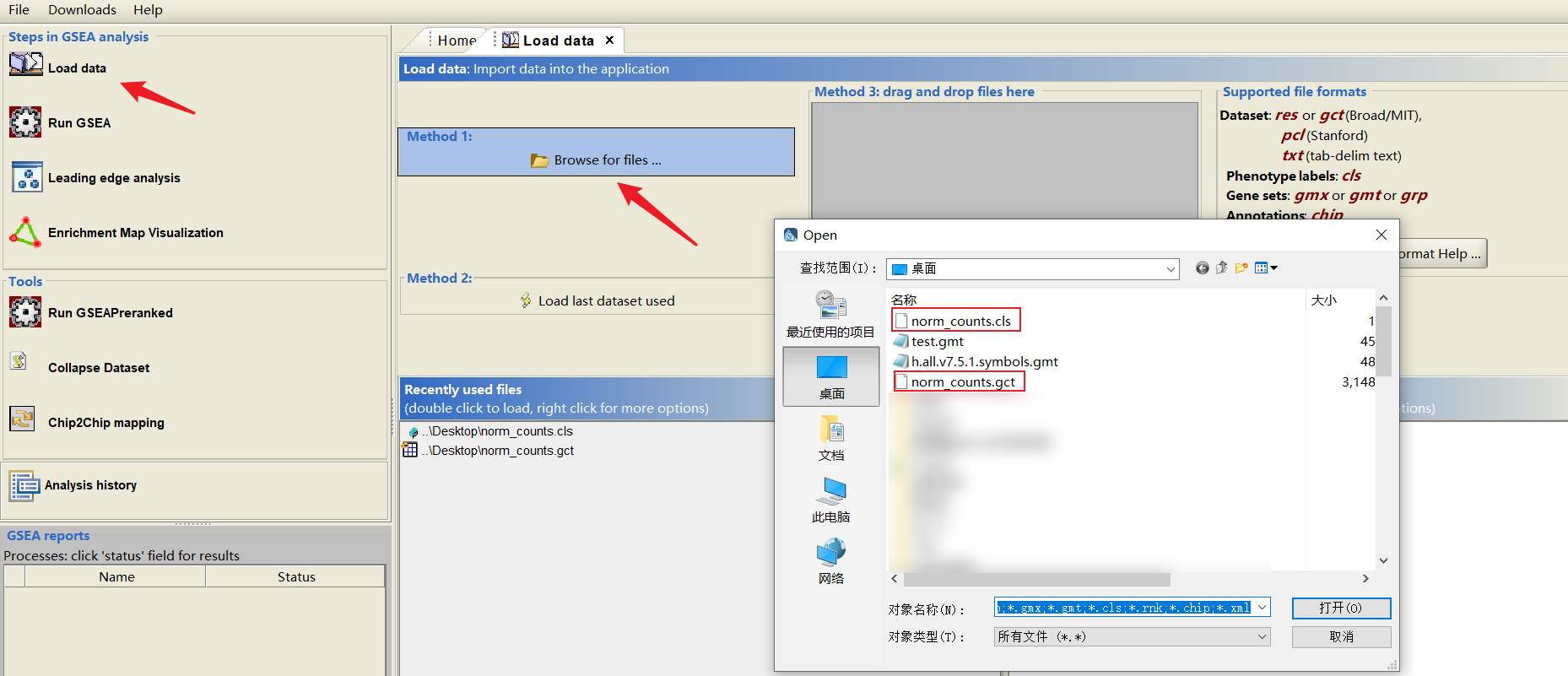
(1)加载数据#
- gct表达矩阵
- cls表型分组信息
- gmt通路基因集(optional)
(2)GSEA分析#
- Expression dataset 选择对应的gct表达矩阵
- Gene sets database 选择工具自提供的msigdb通路集(Hallmark)
- Phenotype labels 选择组间比较方式
- Collapse/Remap to gene symbols 由于已设置gene ID,选择 No_Collapse
- Permutation type 在样本量较少的情况下选择 gene_set
- Analysis name 设置本次GSEA分析的任务名
- 其余参数一般保持默认即可
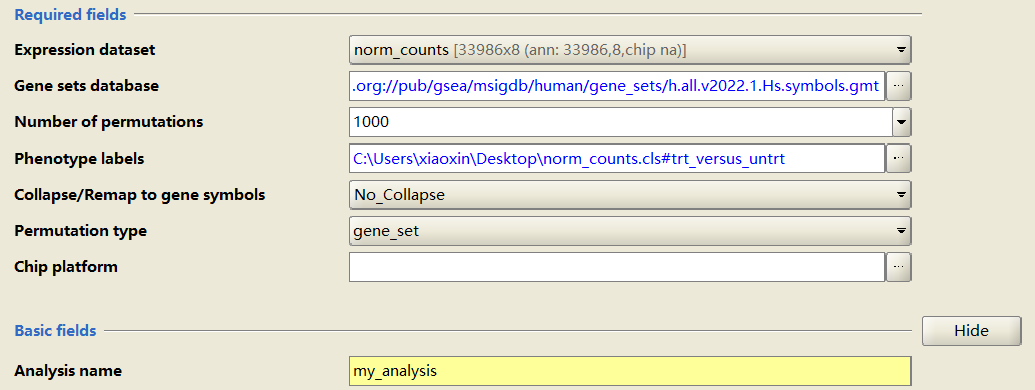
- 最后执行分析Run按钮,等待左侧状态变为Success,点击查看结果。
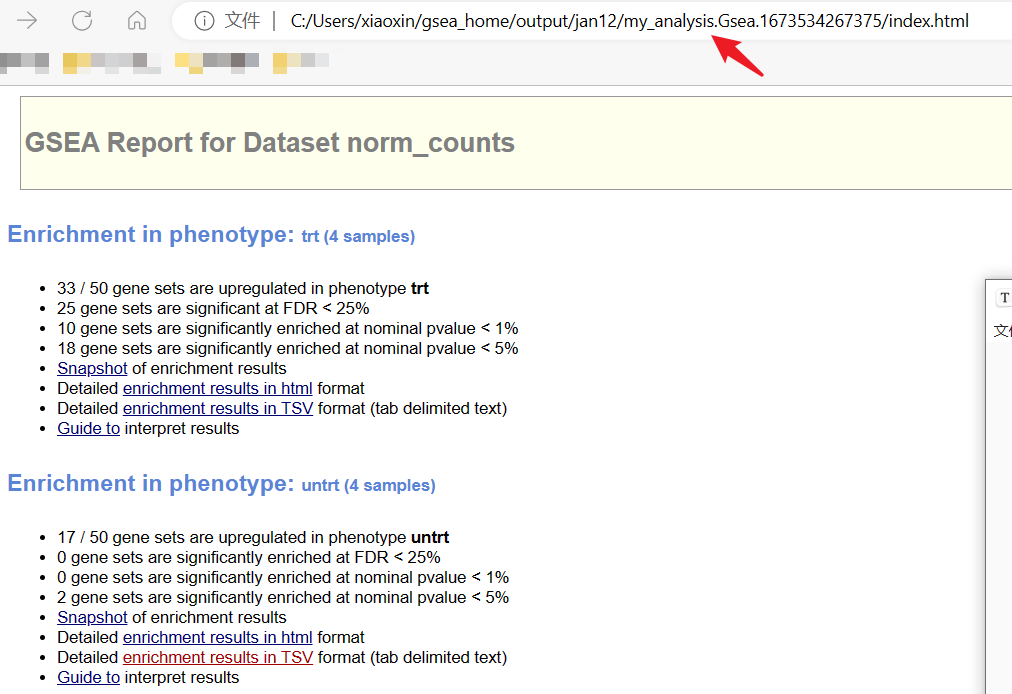
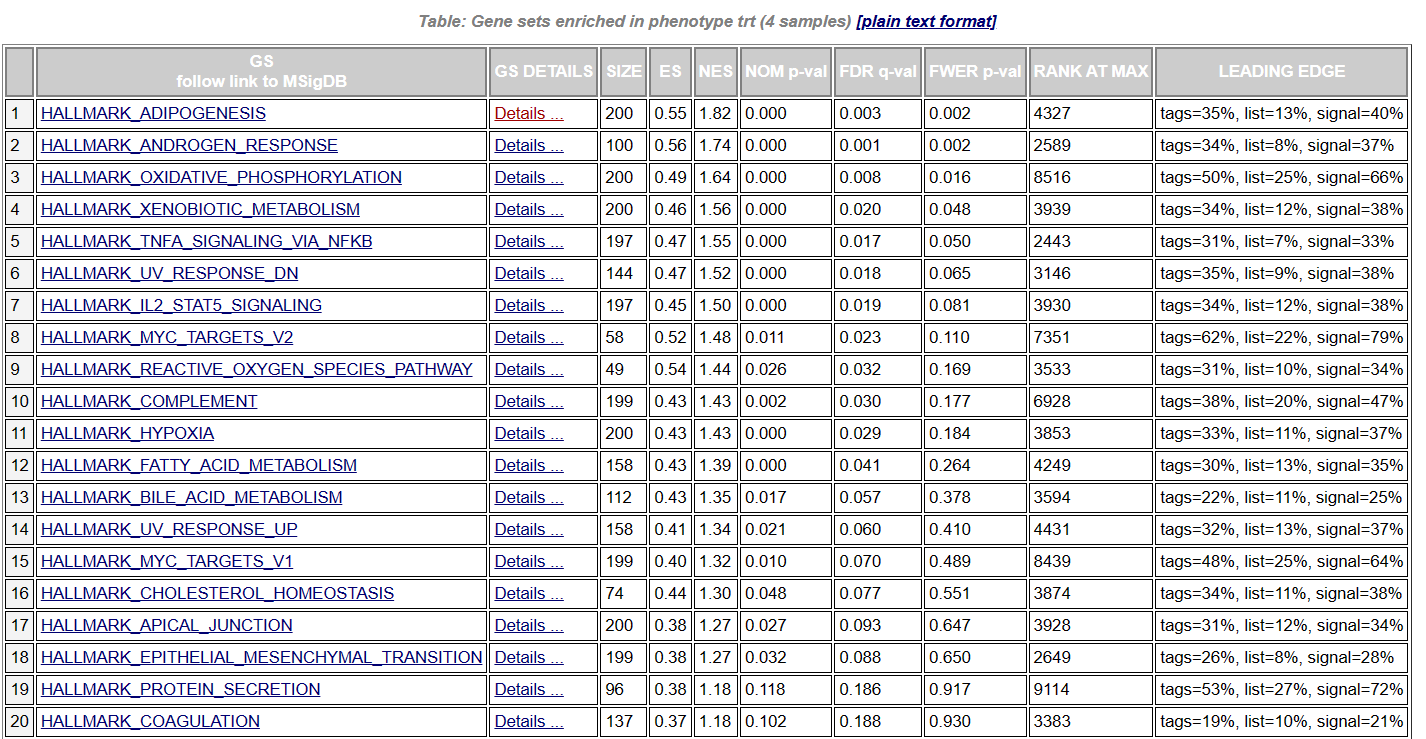
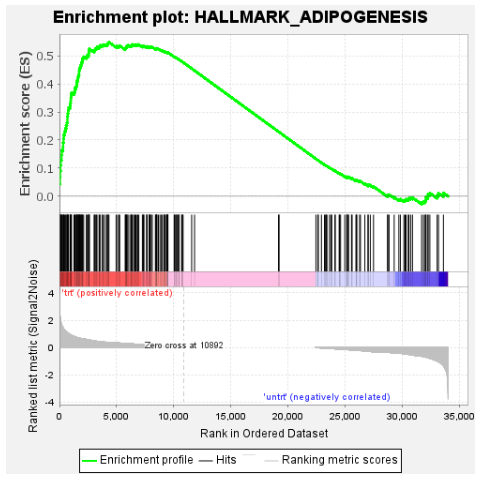
3、分析结果#
- 点击完成状态按钮,进入分析结果报告,可查看每组的相对GSEA富集通路详情与可视化;
- 红色箭头标注的文件地址是本次分析结果所有文件的储存路径,方便查看。