一、基因组大小
(1)人类基因组主要由细胞核的23对染色体组成(核基因组),还包括线粒体中的小DNA分子(线粒体基因组)。
(2)单倍体基因组大概有30亿个碱基对组成,具体到每个染色体的碱基对长度与基因数量如下所示(参照UCSC的hg38)。
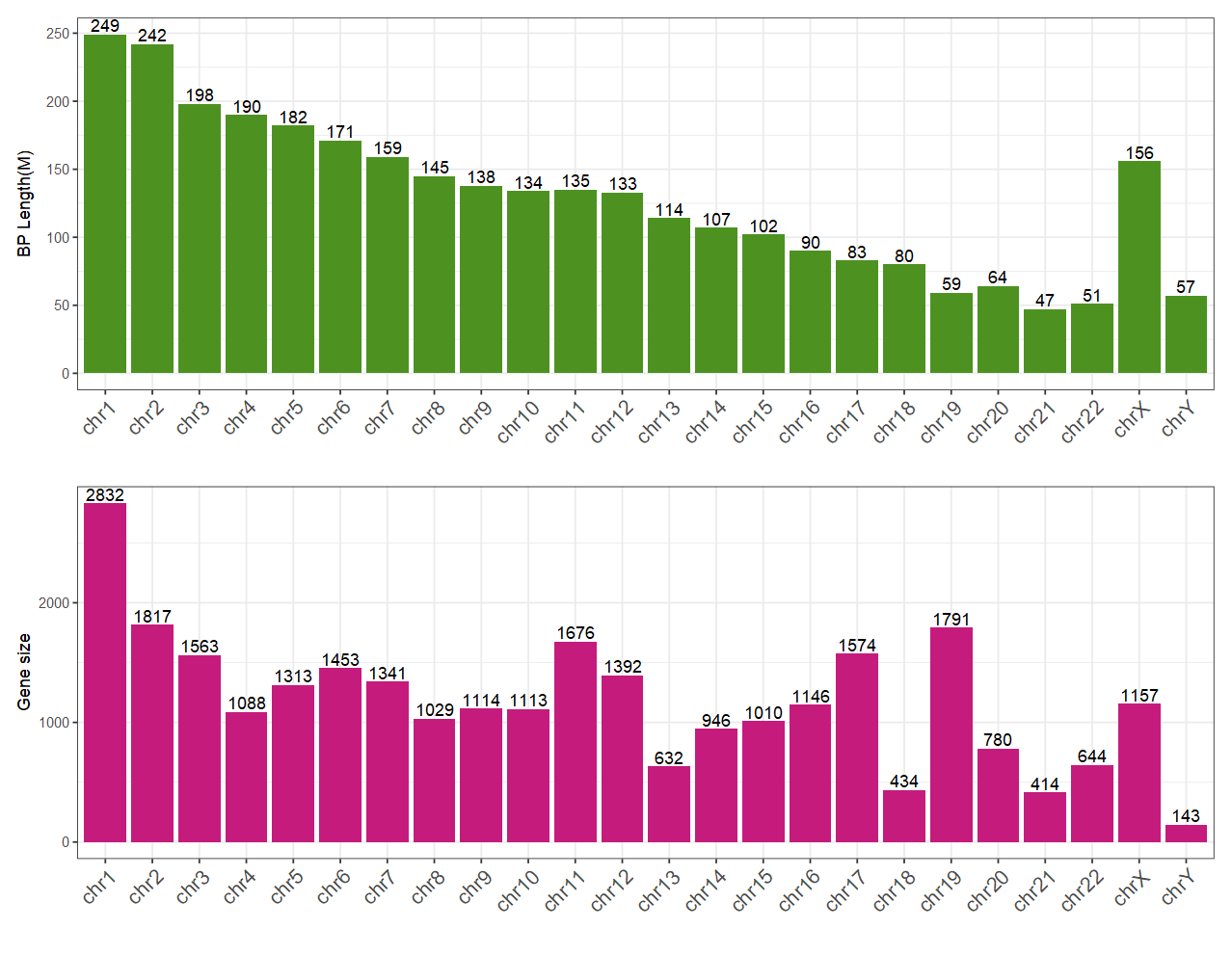
(3)在这30亿个碱基中,仅有1.5%的区域是2~3w个可编码蛋白质的基因。
- 平均基因长度有10Kbp左右,但是不同基因的长度区别很大
- 其余98.5%的区域为非编码区,包括各种调控基因表达的DNA原件,同时也可以转录为non-coding RNA,发挥潜在作用。
二、基因组fasta文件
人类基因组序列信息通常以染色体为单位保存为fasta文件中。
(1)参考基因组一般保存为纯文本格式,即直接记录“A”、“T”、“C”、“G”这样的 ASCII 码字符。而1个 ASCII 字符,大小是 1B,所以,如果按纯文本保存 30亿个字母(单链),就是30亿字母 = 3,000,000,000 B = 3 GB。
(2)理论上应该只有25条序列信息(Chr1:22,X,Y,ChrM)。但是在实际下载的文件中,序列数量远远不止这些。如下以UCSC里的hg38版本为例,有455条序列信息。
除了上述25条序列外,其它序列主要是scaffolds,可大致分为ChrUn, Random, Alt三类。
|
|
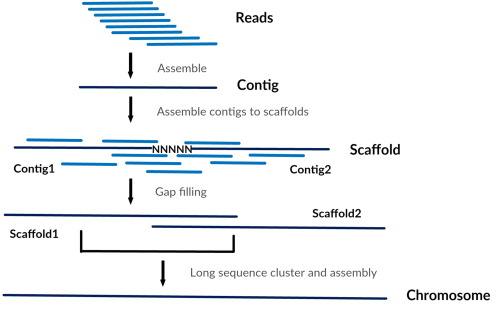
人类基因组主要通过二代测序技术获得,将测得的短Read片段拼接、组装成基因组的染色体序列,需要经历contigs与scaffolds两个过程
-
(1)contigs是依靠read间的重叠拼接的序列(a few kbp long),特点是不含有N碱基;
-
(2)scaffolds则主要依靠read pairs关系进一步拼接contigs,特点是会产生N碱基(a few hundred kbp);
-
(3)最终再由scaffolds拼接成染色体序列。
2.1 Unlocalized scaffolds(*****random)
-
知道这些scaffolds在哪条染色体上,但不知道其在染色体的具体位置及方向
-
format: chr{chromosome number orname}_{sequence_accession}v{sequence_version}_random
|
|
2.2 Unplaced scaffolds(chrUn******)
- 不知道这条scaffolds的所属染色体信息
- format: chrUn_{sequence_accession}v{sequence_version}
|
|
2.3 Alternate loci scaffolds(*****alt)
- 简单理解:参考基因组存在的主要依据是人类99.9%的序列是一致的。但是会存在一些序列在不同人群中不一致。例如49%人群该基因组特定位置为序列A,而49%人群则为序列B,都是正常的。但拿其中一种作为参考基因组都可能不太合适,因此标记出Alternate loci scaffolds。
- format: chr{chromosome number or name}_{sequence_accession}v{sequence_version}_alt
- Alternate loci scaffolds为hg38版本基因组新添类型Sequence,此前hg19版本还没有。
|
|
注意:以上具体的chromosome name均为ucsc的hg版本,与GRCh38略有差异,但基本也是这几种类型sequence。
三、基因结构
1、编码区与非编码区
基因 DNA可分为编码区和非编码区:编码区可转录为 mRNA 并最终翻译成蛋白质;非编码区上具有基因表达的调控元件。
(1)编码区:在转录的过程,从DNA编码区的5’端开始转录生成preRNA,然后进一步加工修饰剪切得到成熟mRNA,进行后续的翻译。
- 外显子:在preRNA 经过剪切或修饰后、被保留的DNA部分。
- 起始密码子(Start Codon):是mRNA上开始合成蛋白质的密码子,也是第一个被核糖体翻译的mRNA密码子。通常为AUG。
- 终止密码子(Stop Codon):代表核糖体翻译的终止。通常为UAG、UAA、UGA。
- UTR(untranslated region):属于外显子部分。即外显子也包含非编码区域。
- 位于5’端的UTR称为5’UTR,如下图橙色标注,从mRNA起点的甲基化鸟嘌呤核苷酸帽延伸至AUG起始密码子。
- 与5’UTR上游的第一个碱基相对应DNA链上的碱基称为TSS(Transcription start sites)
- 位于3’端的UTR称为3’UTR,为mRNA的结束部分,如下图红色标注,从编码区末端的终止密码子延伸至多聚A尾巴(Poly-A)。
- 与3’UTR下游的最后一个碱基相对应DNA链上的碱基称为TTS(Transcription termination sites)
- 位于5’端的UTR称为5’UTR,如下图橙色标注,从mRNA起点的甲基化鸟嘌呤核苷酸帽延伸至AUG起始密码子。
- CDS(Coding sequence):包括mRNA中从5’UTR后的起始密码子开始到3’UTR前的终止密码子的实际编码蛋白序列
- ORF(Open reading frame):从一个起始密码子开始到一个终止密码子结束的一段序列
- 注意与CDS定义上的区别。CDS一定是ORF,ORF不一定是CDS
- Transcript转录本:一条基因通过可变剪切机制可转录形成的一种或多种可供编码蛋白质的成熟的mRNA。
- ORF(Open reading frame):从一个起始密码子开始到一个终止密码子结束的一段序列
- 在最终成熟mRNA的上下游两端修饰有特殊的结构,分别是5’ cap帽子与 3’ Poly-A尾巴。
- Poly-A尾是mRNA’区别于其它non-coding RNA的主要标志

(2)非编码区:位于DNA序列上下游的non-coding区域含有调控基因表达原件
- 启动子Promoter:DNA分子上能与RNA聚合酶结合并形成转录起始复合体的区域,位于TSS位点上游的100~1000碱基序列。
- 真核生物启动子通常会包括一个TATA盒,比较接近TSS区域(50个碱基对以内)
- 终止子Terminator:给RNA聚合酶提供转录终止信号的DNA序列
- 增强子Enhancer:可以蛋白质结合的小段DNA,从而加强所调控基因的转录作用。可以位于靶基因的上游/下游,距离也可以很远。
2、基因组注释文件gtf
- 第一列:染色体ID
- 第三列:region类型
- 第四列:region的起始位点坐标(染色体的第一个碱基序号为1)
- 第五列:region的终止位点坐标
- 第七列:正负链信息(+forward/-reverse)
- 正链:表示实际mRNA序列与坐标对应DNA序列相同,除了(U/T变换)
- 负链:表示实际mRNA序列与坐标对应DNA序列的互补链相同,除了(U/T变换),也需要从5’到3’端读取。
- 第八列:对于实际编码区coding region,表示在这个CDS区域开始编码新密码子的起始index{0,1,2}
- 第九列:region的属性键值对。
|
|
不同平台机构给出的gtf注释文件略有差异,但每列含义基本相同。发现NCBI提供的gff3文件注释信息最全,包括非编码RNA,蛋白质编码RNA、假基因等
|
|
四、下载参考基因组
目前常用的基因组版本为GRCh38/37,hg38/19,前者可通过NCBI/Ensembl下载,后者可通过UCSC网站下载。
如下图所示GRCh38可认为等同于hg38;GRCh37可认为等同于hg19。
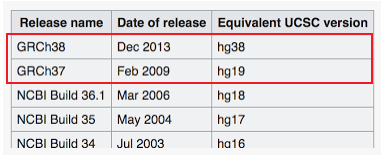
以下载GRCh38/hg38为例,如下
4.1 NCBI
|
|
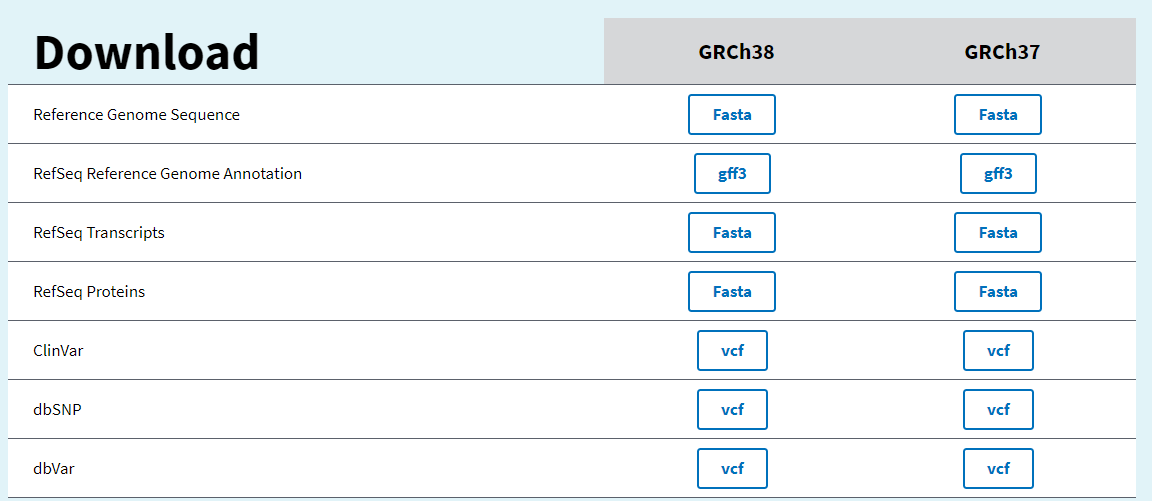
4.2 ensembl
- ensembl的release-103版本可以认为等于GRCh38
- https://asia.ensembl.org/Homo_sapiens/Info/Index
- http://ftp.ensembl.org/pub/release-103/fasta/homo_sapiens/dna/
- https://www.ensembl.org/info/data/ftp/index.html 还包含有cDNA转录本序列
|
|

4.3 UCSC
- http://hgdownload.soe.ucsc.edu/downloads.html
- http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/
|
|
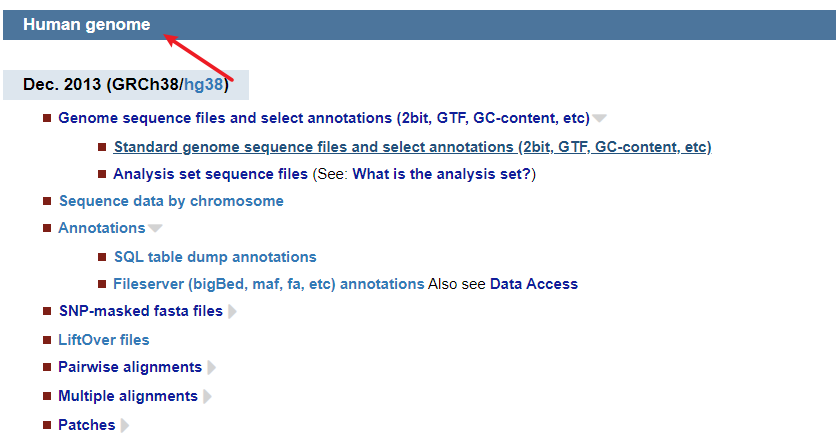
也可以使用之前学习的Refgenie工具下载上述提到的gtf注释文件
五、TxDb与GRanges对象查询gtf
TxDb系列包是将物种基因组的gtf注释信息封装为TxDb对象,以供使用者方便的查询。
|
|
txdb对象支持4种region fearture,共计22种的属性注释。
具体使用时可提供keytypes() 所支持的7种信息之一,查询所匹配的其它(22种内)注释信息。
其中GENEID为Entrez基因ID格式,TXID为Ensemble转录本ID格式
|
|
GenomicFeatures包提供了genes(),transcripts(), exons(), cds(), promoters()函数用于提取特定feature region为GRange对象。
其中可通过filter=list()参数进行筛选符合注释信息的region,可包括
“gene_id”, “tx_id”, “tx_name”, “tx_chrom”, “tx_strand”, “exon_id”, “exon_name”, “exon_chrom”, “exon_strand”, “exon_rank”. “cds_id”, “cds_name”, “cds_chrom”, “cds_strand”
|
|
GRange对象结合BSgenome系列包获取指定feature region的序列信息
|
|
