挖掘GEO数据时,主要一方面是下载GEO的测序数据(包括基因芯片array与RNAseq两类)的表达矩阵。同时会涉及到一些细节问题,例如array芯片ID转换、样本meta信息等。
1、下载GEO数据#
主要使用GEOquery包下载
1.1 array芯片数据#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
|
#BiocManager::install("GEOquery")
library(GEOquery)
gse = getGEO('GSE70213', getGPL = F)
if(length(gse)==1) gse = gse[[1]]
# [1] "ExpressionSet"
# attr(,"package")
# [1] "Biobase"
##(1) 表达矩阵
exp_dat = exprs(gse) %>% as.data.frame()
# GSM1720833 GSM1720834 GSM1720835 GSM1720836
# 10338001 2041.40800 2200.86100 2323.7600 3216.26300
# 10338002 63.78059 65.08438 58.3082 75.86145
# 10338003 635.39040 687.39360 756.0040 1181.92900
# 10338004 251.56680 316.99730 320.5132 592.80600
##(2) 样本信息
meta = pData(gse) %>%
dplyr::select(title, dplyr::ends_with("ch1"))
colnames(meta)=gsub("[:_]ch1", "", colnames(meta))
head(meta[,1:3])
# title source_name organism
# GSM1720833 quad-control-1 quadriceps muscle Mus musculus
# GSM1720834 quad-control-2 quadriceps muscle Mus musculus
# GSM1720835 quad-control-3 quadriceps muscle Mus musculus
# GSM1720836 quad-control-4 quadriceps muscle Mus musculus
# GSM1720837 quad-control-5 quadriceps muscle Mus musculus
# GSM1720838 quad-control-6 quadriceps muscle Mus musculus
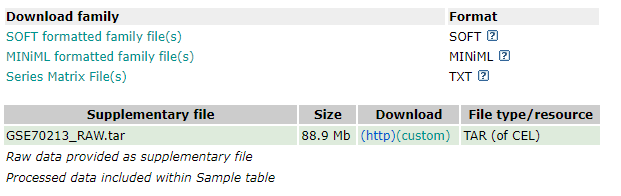
##(3)下载Supplementary file
gse@experimentData@other$supplementary_file
# [1] "ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70213/suppl/GSE70213_RAW.tar"
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
|
##(1) SOFT formatted family file(s)
geo_soft = getGEO(filename = "GSE70213_family.soft")
geo_soft@header #类似上面的gse@experimentData
geo_soft@gsms #每个样本的信息、表达数据
geo_soft@gpls #GPL注释信息
#生成表达矩阵
probesets <- Table(GPLList(geo_soft)[[1]])$ID
exp_dat2 <- do.call('cbind',
lapply(geo_soft@gsms,function(x){
tab <- Table(x)
mymatch <- match(probesets,tab$ID_REF)
return(tab$VALUE[mymatch])
}))
rownames(exp_dat2)=probesets
exp_dat2[1:4,1:4]
##(2) Series Matrix File(s)
series_mt = read.table("GSE70213_series_matrix.txt.gz", comment.char = "!")
#或者查看一下注释的行数
series_mt = data.table::fread("GSE70213_series_matrix.txt.gz",skip = 65,data.table = F)
##(3) cel格式转换,芯片原始数据一般放在Supplementary file中
library(oligo)
CELs = read.celfiles(list.files("GSE70213_RAW/", full.names = T))
expreset = rma(CELs, normalize=F)
exp_dat3 = exprs(expreset)
exp_dat3[1:4,1:4]
colnames(exp_dat3) = stringr::str_split(colnames(exp_dat3), "_", simplify = T)[,1]
|
1.3 RNAseq测序数据#
1
2
3
4
5
6
7
8
9
10
11
12
13
|
##(1) 样本信息
gse = getGEO('GSE87809', getGPL = F)
if(length(gse)==1) gse = gse[[1]]
meta = pData(gse) %>%
dplyr::select(title, dplyr::ends_with("ch1"))
colnames(meta)=gsub("[:_]ch1", "", colnames(meta))
head(meta[,1:3])
##(2) 表达矩阵:因为是RNAseq数据,需要在Series Matrix File(s)或者附件材料里下载。方法同上。
gse@experimentData@other$supplementary_file
# [1] "ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE87nnn/GSE87809/suppl/GSE87809_endometriosis_RNA_seq_FPKM.txt.gz"
|
2、注释芯片ID#
如下一般得到的表达矩阵的基因名还是芯片ID,需要进一步转为基因名。
首先需要下载GPL注释信息,然后整理好对应芯片ID与基因名的关系。根据芯片ID的复杂,可分为若干种情况。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
gse = getGEO('GSE70213', getGPL = F)
if(length(gse)==1) gse = gse[[1]]
# [1] "ExpressionSet"
# attr(,"package")
# [1] "Biobase"
##表达矩阵
exp_dat = exprs(gse) %>% as.data.frame()
# GSM1720833 GSM1720834 GSM1720835 GSM1720836
# 10338001 2041.40800 2200.86100 2323.7600 3216.26300
# 10338002 63.78059 65.08438 58.3082 75.86145
# 10338003 635.39040 687.39360 756.0040 1181.92900
# 10338004 251.56680 316.99730 320.5132 592.80600
##获取GPL平台
gse@annotation
# [1] "GPL6246"
##下载GPL注释信息
GPL=getGEO("GPL6246")
GPL@dataTable@table[1:4,]
dim(GPL@dataTable@table)
# [1] 35557 12
|
查看下载得到的GPL注释信息,可分为如下几种情况
2.1 直接注释有Symbol基因名#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
GPL=getGEO("GPL96")
anno=GPL@dataTable@table
colnames(anno)
#一般probe ID都在第一列
probe2symbol=anno[,c(1,grep("symbol",colnames(anno),ignore.case = T))]
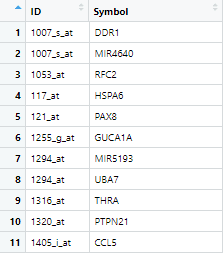
head(probe2symbol)
# ID Gene Symbol
#1 1007_s_at DDR1 /// MIR4640
#2 1053_at RFC2
#3 117_at HSPA6
#4 121_at PAX8
#5 1255_g_at GUCA1A
#6 1294_at MIR5193 /// UBA7
colnames(probe2symbol)=c("ID","Symbol")
|
1
2
3
4
5
6
7
8
9
10
|
#去掉未匹配到的情况
probe2symbol=probe2symbol[!(probe2symbol[,2] %in% c("",NA,"---")),]
#针对一个探针匹配到多种gene的情况
tmp=unlist(lapply(1:nrow(probe2symbol),function(i){
gene=trimws(unlist(strsplit(probe2symbol[,2][i],"//*")))
names(gene)=rep(probe2symbol[,1][i],length(gene))
return(gene)
}))
probe2symbol=data.frame(ID=names(tmp),Symbol=tmp)
|
2.2 注释有其它格式基因名#
这种情况需要知道不同格式基因名转换的方法,以下举了两个例子。
(1)提供的是ENTREZ_GENE_ID 格式
- 例如平台
GPL17889

1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db)
GPL=getGEO("GPL17889")
anno=GPL@dataTable@table
anno[,2]=as.character(anno[,2]) #因为ENTREZID是纯数字组成,需要转为字符型
gene_bridge<-AnnotationDbi::select(org.Hs.eg.db, keys=anno[,2],
columns=c("SYMBOL"), #目标格式
keytype="ENTREZID") #当前格式
probe2symbol=dplyr::left_join(anno, gene_bridge, by=c("ORF"="ENTREZID"))
head(probe2symbol)
# ID ORF SYMBOL
# 1 1_at 1 A1BG
# 2 10_at 10 NAT2
# 3 100_at 100 ADA
# 4 1000_at 1000 CDH2
# 5 10000_at 10000 AKT3
# 6 100009613_at 100009613 LINC02584
probe2symbol=probe2symbol[,-2]
colnames(probe2symbol)=c("ID","Symbol")
|
(2)提供的是NCBI Accession Number格式
- 例如平台
GPL16686,类似NR_046018、NM_152486、AA972198、BX101169格式

- 同样利用
org.Hs.eg.db包转换
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
GPL=getGEO("GPL16686")
anno=GPL@dataTable@table
probe2symbol=anno[,c(1,6)]
probe2symbol=probe2symbol[probe2symbol[,2]!="",]
gene_bridge=AnnotationDbi::select(org.Hs.eg.db, keys=probe2symbol[,2],
columns=c("SYMBOL"), #目标格式
keytype="ACCNUM") #当前格式
probe2symbol=dplyr::left_join(probe2symbol,gene_bridge,by=c("GB_ACC"="ACCNUM"))
# ID GB_ACC SYMBOL
# 1 16657436 NR_046018 DDX11L1
# 2 16657450 NR_024368 LINC01000
# 3 16657450 NR_024368 LINC01000
# 4 16657450 NR_024368 LINC01000
# 5 16657476 NR_029406 RPL23AP87
# 6 16657476 NR_029406 RPL23AP87
probe2symbol=probe2symbol[,-2]
colnames(probe2symbol)=c("ID","Symbol")
probe2symbol=probe2symbol[!(probe2symbol[,2] %in% c("",NA,"---")),]
|
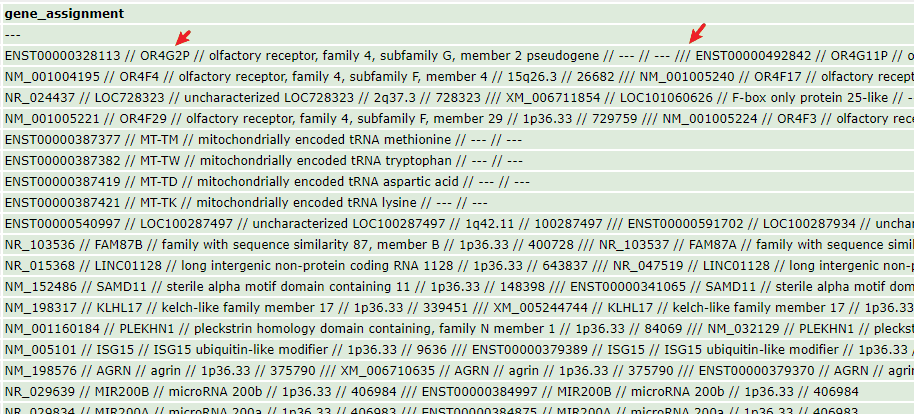
2.3 gene_assignment的特殊注释#
- 例如GPL6244,如下图解释(可以看到也可以是用
GB_list进行基因ID转换)

- 可以看到当一个探针匹配到多个基因时,是用
///分隔;用//分隔同一基因的不同角度注释,其中Symbol ID一般位于第二条信息。需要用一些R语言技巧提取出来。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
GPL=getGEO("GPL6244")
anno=GPL@dataTable@table
anno=anno[anno[,"gene_assignment"]!="---",]
tmp=unlist(lapply(1:nrow(anno),function(i){
tmp1=strsplit(anno[,"gene_assignment"][i],"///") #1个探针对多个基因的拆分
tmp2=unique(trimws(stringr::str_split(unlist(tmp1),"//",simplify = T)[,2])) #提取第二个symbol ID
names(tmp2)=rep(anno[,1][i],length(tmp2))
return(tmp2)
}))
probe2symbol=data.frame(ID=names(tmp),Symbol=tmp)
# ID Symbol
# 1 7896738 OR4G2P
# 2 7896738 OR4G11P
# 3 7896738 OR4G1P
# 4 7896740 OR4F4
# 5 7896740 OR4F17
# 6 7896740 OR4F5
|
2.4 AnnoProbe包#
- AnnoProbe是生信技能树曾老师团队写的一个包,整理有常见芯片平台与基因名的对应关系。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
#devtools::install_github("jmzeng1314/AnnoProbe")
library(AnnoProbe)
AnnoProbe::getGPLList() %>% dim()
# [1] 185 2
AnnoProbe::idmap(gpl = "GPL570") %>% head()
# trying URL 'http://49.235.27.111/GEOmirror/GPL/GPL570_bioc.rda'
# Content type 'application/octet-stream' length 264292 bytes (258 KB)
# downloaded 258 KB
#
# file downloaded in C:/Users/xiaoxin/Desktop/GEOquery
# probe_id symbol
# 193731 1053_at RFC2
# 193732 117_at HSPA6
# 193733 121_at PAX8
# 193734 1255_g_at GUCA1A
# 193735 1316_at THRA
# 193736 1320_at PTPN21
|
Plus:GEO搜索方式#
1
2
3
4
5
6
7
8
9
10
11
|
# https://www.ncbi.nlm.nih.gov/gds
"blood"[All Fields] AND
"Homo Sapiens"[porgn] AND
("Alzheimer"[All Fields]) AND
("gds"[Filter] OR "gse"[Filter] OR "gsm"[Filter]) AND
("Expression profiling by array"[Filter] OR "Expression profiling by high throughput sequencing"[Filter])
# "Homo Sapiens"[porgn] AND
# "blood"[All Fields] AND
|