TCGAbiolinks包是一站式分析TCGA数据的R包工具,它集成了TCGA数据下载、分析、可视化的全部流程。此次系列笔记主要跟着 TCGAbiolinks帮助文档重新学习下TCGA数据挖掘流程。
官方文档:https://bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/index.html
文献:TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data https://pubmed.ncbi.nlm.nih.gov/26704973/
安装包
1 2 3 4 5 6 7if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") #From github BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data") BiocManager::install("BioinformaticsFMRP/TCGAbiolinks") #From Bioconductor BiocManager::install("TCGAbiolinks")
一、查找TCGA数据
- 主要通过
GDCquery()函数查找数据
|
|
1 选择肿瘤类型
project参数:指定一个或多个感兴趣的TCGA项目名- 如下代码所示,供包括33种TCGA癌症类型。癌症简称对应全称及中文名见文末。
|
|
2 hg19/hg38
- 主要根据参考基因组的不同,包含两套数据:GDC Legacy Archive【主要GRCh37 (hg19)】,GDC harmonized database【GRCh38 (hg38)】
- 通过设置参数
legacy,默认为FALSE(hg38);TRUE则表示使用hg19参考基因组的测序数据。

3 下载数据类型
基于上述的参数,我们可以设置如下参数,交代我们的目标数据类型
data.category =指定下载什么类型的数据:如转录组数据、临床数据….
|
|
data.type =更加细节的数据类型选择,例如harmonized的Transcriptome Profiling有Gene Expression Quantification,Isoform Expression Quantification,miRNA Expression Quantification,Splice Junction Quantification4种数据。而我们一般需要的是Gene Expression Quantification。workflow.type =同一个测序数据可能有不同的pipeline处理流程,如果有不同的处理流程,需要选择。据最新的TCGA版本,仅提供STAR - Counts。platform =测序平台(optional)file.type =具体的数据文件(optional, for legacy) 如果不知道目标数据的上述信息,可以参考下面的概述
具体参考文末第二点。
4 样本标签Barcode
完整的barcode:形如 TCGA-G4-6317-02A-11D-2064-05,这个标签包含了从病人来源到测序过程、分析的所有信息,如下图所示比较重要的是Participant、Sample 、Portion三个部分,分别交代了病人编号、样本类型、测序类型
病人的id:形如 TCGA-G4-6317
样本来源的id:形如 TCGA-G4-6317-02
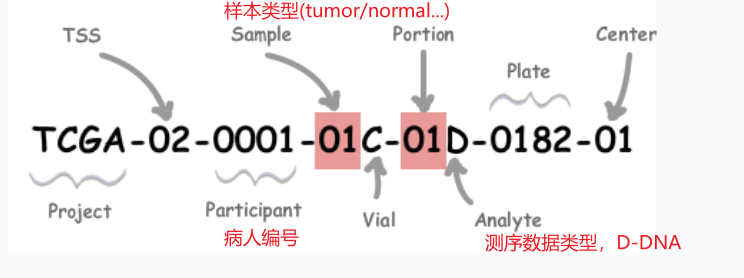
-
其中比较重要的是交代样本类型的
Sample的两位数信息,是后面进行差异分析的分组依据。具体对应的含义如下。例如01表示病人的原位瘤组织;11表示来自病人的正常组织….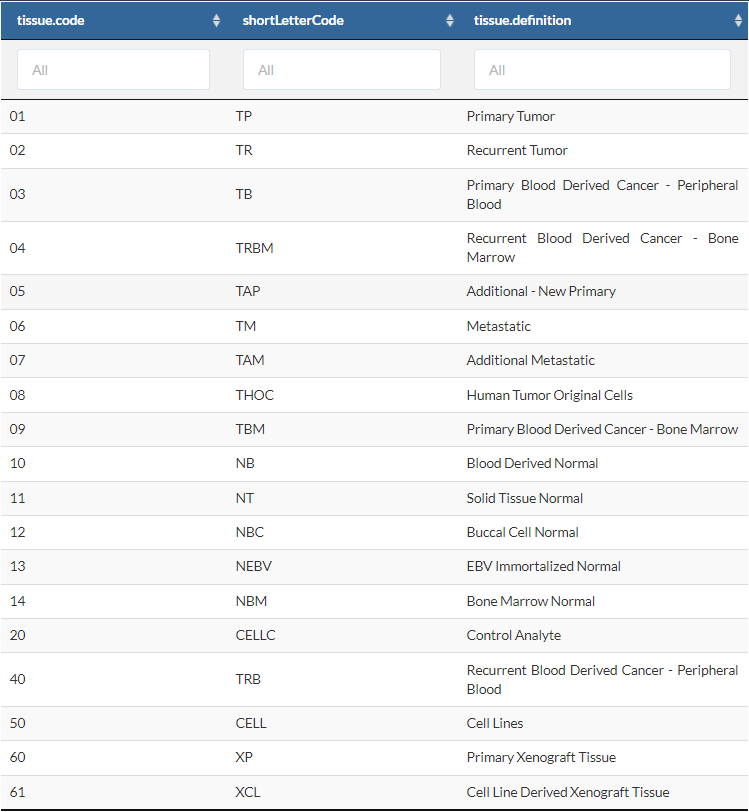
-
基于上述理解,我们也可以设置
sample.type =参数指定下载感兴趣的样本类型数据,例如sample.type = "Primary Tumor" -
对于给定的TCGA barcode,可以利用
TCGAquery_SampleTypes()提取出目标分组的样本;TCGAquery_MatchedCoupledSampleTypes()函数可以提取来自同一病人的配对样本数据。
|
|
如上是查询TCGA目标数据的几种常见标准,还有几个参数没有介绍,可参看函数帮助文档。可根据自己的目的灵活设置上述参数。
二、从数据下载到差异分析
以胆管癌为例,演示从数据下载到差异分析的全流程。
1 Legacy(hg19)
1.1 下载数据
|
|
1.2 探索数据
|
|
1.3 整理数据
|
|
1.4 差异分析
|
|
2 Harmonized(hg38)
2.1 下载数据
|
|
2.2 探索数据
|
|
2.3 整理数据
|
|
2.4 差异分析
(1) edgeR
|
|
(2) Deseq2
|
|
四、关于病人的临床数据与肿瘤分型
1、获取病人的临床数据
- 如上在
GDCprepare()过程中,会自动注释病人样本的临床信息。 - 我们也可以预先单独下载每个病人的临床数据,以供参考。
方法一:GDCquery() pipeline
|
|
方法二:GDCquery_clinic()
- 根据官方介绍,这个函数下载的是indexed clinical: a refined clinical data that is created using the XML files(方法一).
- 这种方法下载速度较快,建议优先使用。如果没有想要的信息,再使用方法一。
|
|
2、获取病人的肿瘤分型
PanCancerAtlas_subtypes()The columns “Subtype_Selected” was selected as most prominent subtype classification (from the other columns)
|
|
TCGAquery_subtype()These subtypes will be automatically added in the summarizedExperiment object through GDCprepare. But you can also use the TCGAquery_subtype function to retrieve this information.
|
|
GDCquery_Maf()函数可以支持下载突变数据,这里就暂时不学习了。之后有机会再了解一下。
补充
1 、TCGA简称对应全称以及中文名
| Study Abbreviation | Study Name | 中文名 |
|---|---|---|
| ACC | Adrenocortical carcinoma | 肾上腺皮质癌 |
| BLCA | Bladder Urothelial Carcinoma | 膀胱尿路上皮癌 |
| BRCA | Breast invasive carcinoma | 浸润性乳腺癌 |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma | 宫颈鳞状细胞癌和宫颈内腺癌 |
| CHOL | Cholangiocarcinoma | 胆管癌 |
| COAD | Colon adenocarcinoma | 结肠腺癌 |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma | 淋巴样肿瘤弥漫大b细胞淋巴瘤 |
| ESCA | Esophageal carcinoma | 食管癌癌 |
| GBM | Glioblastoma multiforme | 多形性成胶质细胞瘤 |
| HNSC | Head and Neck squamous cell carcinoma | 头颈部鳞状细胞癌 |
| KICH | Kidney Chromophobe | 肾嫌色细胞癌 |
| KIRC | Kidney renal clear cell carcinoma | 肾透明细胞癌 |
| KIRP | Kidney renal papillary cell carcinoma | 肾乳头状细胞癌 |
| LAML | Acute Myeloid Leukemia | 急性髓系白血病 |
| LGG | Brain Lower Grade Glioma | 脑低级别胶质瘤 |
| LIHC | Liver hepatocellular carcinoma | 肝脏肝细胞癌 |
| LUAD | Lung adenocarcinoma | 肺腺癌 |
| LUSC | Lung squamous cell carcinoma | 肺鳞癌 |
| MESO | Mesothelioma | 间皮瘤 |
| OV | Ovarian serous cystadenocarcinoma | 卵巢浆液性囊腺癌 |
| PAAD | Pancreatic adenocarcinoma | 胰腺腺癌 |
| PCPG | Pheochromocytoma and Paraganglioma | 嗜铬细胞瘤和副神经节瘤 |
| PRAD | Prostate adenocarcinoma | 前列腺腺癌 |
| READ | Rectum adenocarcinoma | 直肠腺癌 |
| SARC | Sarcoma | 肉瘤 |
| SKCM | Skin Cutaneous Melanoma | 皮肤皮肤黑色素瘤 |
| STAD | Stomach adenocarcinoma | 胃腺癌 |
| TGCT | Testicular Germ Cell Tumors | 睾丸生殖细胞肿瘤 |
| THCA | Thyroid carcinoma | 甲状腺癌 |
| THYM | Thymoma | 胸腺瘤 |
| UCEC | Uterine Corpus Endometrial Carcinoma | 子宫内膜癌 |
| UCS | Uterine Carcinosarcoma | 子宫癌肉瘤 |
| UVM | Uveal Melanoma | 葡萄膜黑色素瘤 |
2 、GDC所有数据类型
2.1 GDC harmonized database
| Data.category | Data.type | Workflow.Type | Platform |
|---|---|---|---|
| Transcriptome Profiling | Gene Expression Quantification | HTSeq - Counts | |
| Transcriptome Profiling | Gene Expression Quantification | HTSeq - FPKM | |
| Transcriptome Profiling | Gene Expression Quantification | HTSeq - FPKM-UQ | |
| Transcriptome Profiling | Gene Expression Quantification | STAR - Counts | |
| Transcriptome Profiling | Isoform Expression Quantification | - | |
| Transcriptome Profiling | miRNA Expression Quantification | - | |
| Transcriptome Profiling | Splice Junction Quantification | ||
| Copy number variation | Copy Number Segment | ||
| Copy number variation | Masked Copy Number Segment | ||
| Copy number variation | Gene Level Copy Number Scores | ||
| Simple Nucleotide Variation | Masked Somatic Mutation | MuSE Variant Aggregation and Masking | |
| Simple Nucleotide Variation | Masked Somatic Mutation | MuTect2 Variant Aggregation and Masking | |
| Simple Nucleotide Variation | Masked Somatic Mutation | SomaticSniper Variant Aggregation and Masking | |
| Simple Nucleotide Variation | Masked Somatic Mutation | VarScan2 Variant Aggregation and Masking | |
| Raw Sequencing Data | - | ||
| Biospecimen | Slide Image | ||
| Biospecimen | Biospecimen Supplement | ||
| Clinical | - | ||
| DNA Methylation | Methylation Beta Value | Illumina Human Methylation 450 | |
| DNA Methylation | Methylation Beta Value | Illumina Human Methylation 27 |
2.2 GDC Legacy Archive
| Data.category | Data.type | Platform | file.type |
|---|---|---|---|
| Copy number variation | - | Affymetrix SNP Array 6.0 | nocnv_hg18.seg |
| Copy number variation | - | Affymetrix SNP Array 6.0 | hg18.seg |
| Copy number variation | - | Affymetrix SNP Array 6.0 | nocnv_hg19.seg |
| Copy number variation | - | Affymetrix SNP Array 6.0 | hg19.seg |
| Copy number variation | - | Illumina HiSeq | - |
| Simple nucleotide variation | Simple somatic mutation | ||
| Raw sequencing data | |||
| Biospecimen | |||
| Clinical | |||
| Protein expression | MDA RPPA Core | - | |
| Gene expression | Gene expression quantification | Illumina HiSeq | normalized_results |
| Gene expression | Gene expression quantification | Illumina HiSeq | results |
| Gene expression | Gene expression quantification | HT_HG-U133A | - |
| Gene expression | Gene expression quantification | AgilentG4502A_07_2 | - |
| Gene expression | Gene expression quantification | AgilentG4502A_07_1 | - |
| Gene expression | Gene expression quantification | HuEx-1_0-st-v2 | FIRMA.txt |
| Gene expression | Gene expression quantification | gene.txt | |
| Gene expression | Isoform expression quantification | - | - |
| Gene expression | miRNA gene quantification | - | hg19.mirna |
| Gene expression | miRNA gene quantification | hg19.mirbase20 | |
| Gene expression | miRNA gene quantification | mirna | |
| Gene expression | Exon junction quantification | - | - |
| Gene expression | Exon quantification | - | - |
| Gene expression | miRNA isoform quantification | - | hg19.isoform |
| Gene expression | miRNA isoform quantification | - | isoform |
| DNA methylation | Illumina Human Methylation 450 | Not used | |
| DNA methylation | Illumina Human Methylation 27 | Not used | |
| DNA methylation | Illumina DNA Methylation OMA003 CPI | Not used | |
| DNA methylation | Illumina DNA Methylation OMA002 CPI | Not used | |
| DNA methylation | Illumina Hi Seq | ||
| DNA methylation | Bisulfite sequence alignment | ||
| DNA methylation | Methylation percentage | ||
| DNA methylation | Aligned reads | ||
| Raw microarray data | Raw intensities | Illumina Human Methylation 450 | idat |
| Raw Microarray Data | Raw intensities | Illumina Human Methylation 27 | idat |
| Structural Rearrangement | |||
| Other |