网站:https://cellxgene.cziscience.com/
API:https://chanzuckerberg.github.io/cellxgene-census/
Schema:https://github.com/chanzuckerberg/single-cell-curation/blob/main/schema/3.0.0/schema.md
CELLxGENE 是一套工具,可帮助科学家查找、下载、探索、分析、注释和发布单细胞数据集。它包含几个功能强大的工具,具有各种功能,可帮助您处理单细胞数据。
Chan Zuckerberg Initiative(CZI)是由马克·扎克伯格(Mark Zuckerberg)和妻子普莉希拉·陈(Priscilla Chan)于2015年创立的慈善组织。其使命是通过支持科学、教育和社区发展来推动人类潜能和促进平等。
其核心是整合了大量的单细胞转录组数据集于一体,具有如下特点
- 物种来源主要是Homo sapiens, Mus musculus;
- 样本主要来自大多数组织的正常细胞(Normal, 即non-disease);
- 数据类型是未标准化的原始Counts表达数据;
- 在目前最新的版本(20240701)中,细胞总量已分别达到74M(human), 41M(mouse)
Note that the Census RNA data includes duplicate cells present across multiple datasets. Duplicate cells can be filtered in or out using the cell metadata variable
is_primary_datawhich is described in the Census schema.
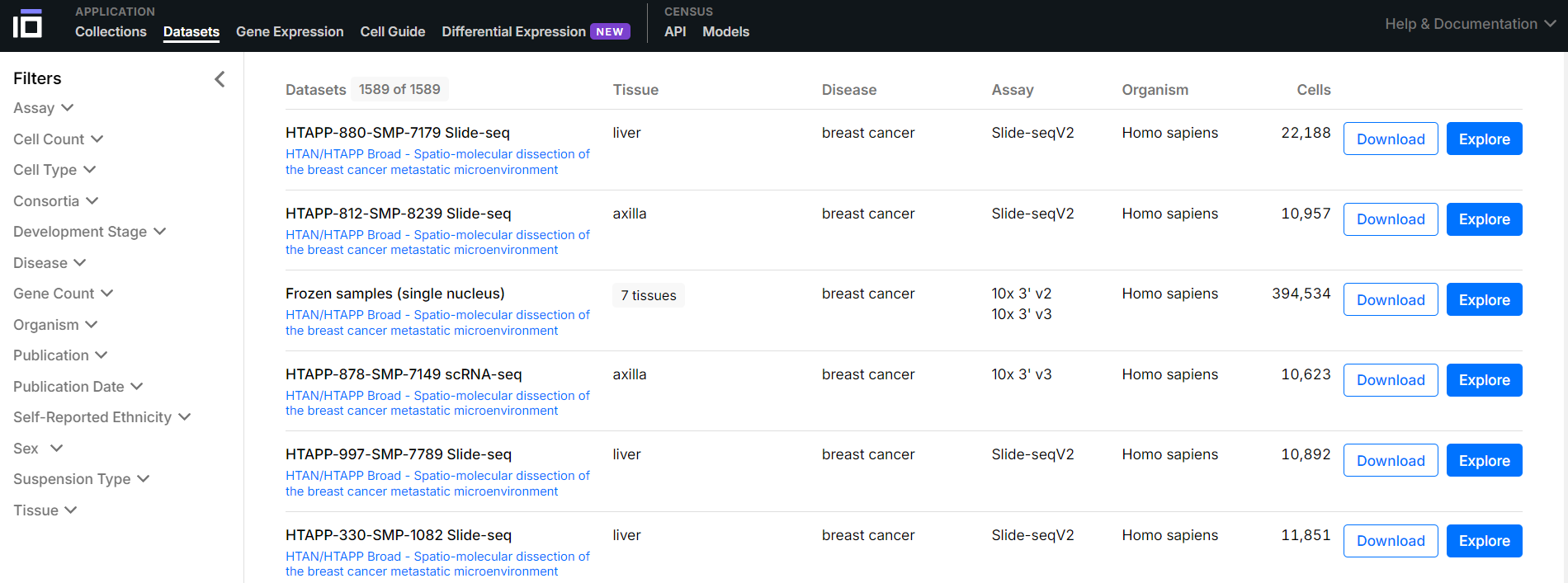
CELLxGENE基于这些数据资源主要提供两类功能,Download (下载) & Explore (分析)。下面将主要学习下数据获取的方式。
方式1(如上图):从网站的Datasets页面,根据提供的多种filter筛选目标数据集,然后点击下载按钮;提供了h5ad与rds两种格式,可分别用于Python的scanpy(v0.8)分析以及R的Seurat分析(v5)
方式2:使用网站提供的API接口进行定制化下载,如下主要记录对于Python端的简单学习。
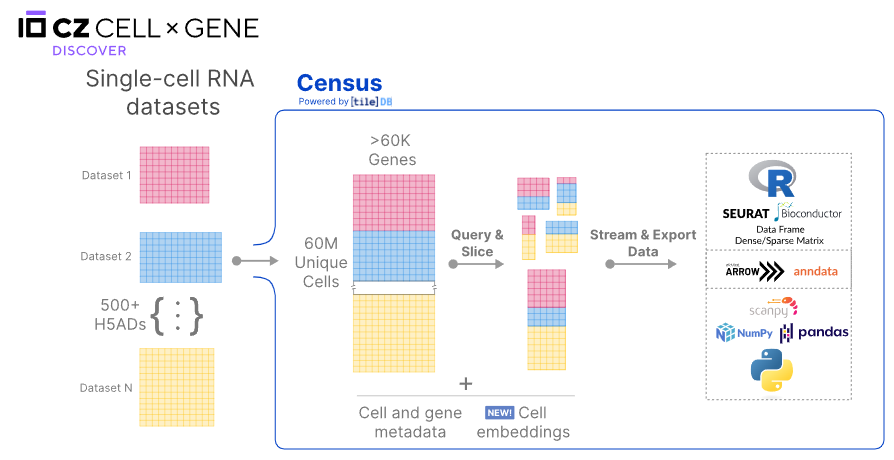
简单来说,可以将census视为CELLxGENE将所有细胞数据进行的整合。我们可以基于特定的cell或者gene metadata条件(Slice),筛选目标的单细胞数据。
|
|
- 查看所有的cell metadata类型(2023-05-15版本)
|
|
- 获取该版本所有细胞的metadata(建议使用特定条件筛选,这里仅为了下面探索全部的类别信息)
|
|
详见:https://github.com/lishensuo/utils/tree/main/CELLxGene/census_20230515
- 获取目前最新版本(20240701)所有细胞的metadata
|
|
详见:https://github.com/lishensuo/utils/tree/main/CELLxGene/census_20240701
- 下面简单探索前列腺细胞(Prostate gland cells)的metadata
|
|
- 单细胞数据下载,以上面前列腺组织的fibroblast (669 cells)为例
|
|
- 另外的下载方式(scGPT采用)
- 首先查询目标细胞的soma_joinid数据
- 然后直接根据上述ID,获取对应细胞的表达数据
|
|
API的下载速度目前体验下来,比较不稳定,有时非常慢。在时间允许的情况下,可以编写脚本,在后台慢慢下载。