ShinyCell包是由杜克-新加坡国立大学医学院的John F. Ouyang团队开发的单细胞分析工具包,实现基于shiny网页交互式展示单细胞数据;于2021年3月发表于Bioinformatics杂志。如文章中介绍,ShinyCell相比同类工具具有多个优势,例如直观的side-by-side的降维可视化方式,hdf5格式保存表达矩阵从而读取快速,支持pdf/png保存图片,支持多种常见单细胞数据类型等。参考其教程文档,学习记录如下。
1. 安装R包#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
# (1) 安装ShinyCell所需的依赖包
reqPkg = c("data.table", "Matrix", "hdf5r", "reticulate", "ggplot2",
"gridExtra", "glue", "readr", "RColorBrewer", "R.utils", "Seurat")
newPkg = reqPkg[!(reqPkg %in% installed.packages()[,"Package"])]
if(length(newPkg)){install.packages(newPkg)}
# (2) 安装运行shiny所需的R包
reqPkg = c("shiny", "shinyhelper", "data.table", "Matrix", "DT", "hdf5r",
"reticulate", "ggplot2", "gridExtra", "magrittr", "ggdendro")
newPkg = reqPkg[!(reqPkg %in% installed.packages()[,"Package"])]
if(length(newPkg)){install.packages(newPkg)}
# (3) 最后安装ShinyCell包
# github直接安装
devtools::install_github("SGDDNB/ShinyCell")
# 源文件本地安装
devtools::install_local("ShinyCell-master.zip")
|
- ShinyCell支持多种单细胞数据类型,包括Seurat, SCE(singlecellexperiment), h5ad, loom;并均提供了相应的示例文件;
- 如其文档所强调,ShinyCell是一个可视化工具,而不是分析工具;所以提供的单细胞数据需要已经完成基础的上游分析;
- 下面主要以Seurat类型为例进行学习
1
2
3
|
library(Seurat)
library(ShinyCell)
library(shiny)
|
2. 基础用法#
2.1 示例数据
1
2
3
|
# getExampleData() # ~200MB
# http://files.ddnetbio.com/hrpiFiles/readySeu_rset.rds
seu = readRDS("readySeu_rset.rds")
|
单细胞数据里需包括(1)标准化表达矩阵;(2)细胞meta信息;(3)降维信息。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
|
### (1)标准化表达矩阵
dim(seu)
# [1] 12590 9852
Assays(seu)
# [1] "RNA"
seu@assays$RNA@data[1:4,1:4] #注意是ENSEMBL基因ID
# 4 x 4 sparse Matrix of class "dgCMatrix"
# FM_AAACCTGAGAAACGCC-1 FM_AAACCTGAGTGGAGTC-1 FM_AAACCTGCATAGAAAC-1 FM_AAACCTGTCATTCACT-1
# ENSG00000000003 . . 0.8413237 .
# ENSG00000000419 . . . .
# ENSG00000000457 . . . .
# ENSG00000000460 . . . .
### (2)细胞meta信息
head(t(seu@meta.data[1,]),10)
# FM_AAACCTGAGAAACGCC-1
# orig.ident "FM"
# RNA_snn_res.0.5 "5"
# library "FM"
# nUMI "13798"
# nGene "3522"
# pctMT "1.232063"
# pctHK "20.77113"
# score_S "-0.2176265"
# score_G2M "-0.3646008"
# phase "G1"
### (3)降维信息
names(seu@reductions)
# [1] "pca" "tsne" "umap" "diffmap" "fdl"
head(Embeddings(seu, reduction = "umap"))
# UMAP_1 UMAP_2
# FM_AAACCTGAGAAACGCC-1 -7.728941 -3.2370806
# FM_AAACCTGAGTGGAGTC-1 -7.048894 2.8379904
# FM_AAACCTGCATAGAAAC-1 -7.768705 1.6628623
# FM_AAACCTGTCATTCACT-1 -6.059119 3.3121483
# FM_AAACCTGTCTCTGAGA-1 -8.030442 1.5388506
# FM_AAACGGGCAGGCGATA-1 -9.831629 -0.5126803
|
2.2 生成shiny网页
(1)生成meta配置文件
1
2
3
4
5
6
7
8
9
10
11
12
|
scConf = createConfig(seu)
head(scConf) # 每行对应一项meta的title、level name、颜色方案等信息
t(scConf[1,])
# [,1]
# ID "orig.ident"
# UI "orig.ident"
# fID "FM|NR|PR|RR"
# fUI "FM|NR|PR|RR"
# fCL "#A6CEE3|#B89B74|#ED8F47|#B15928"
# fRow "1"
# default "1"
# grp "TRUE"
|
ID, fID 对应原始数据;UI, fUI, fCL 分布表示在shiny中展示的meta title,level name,colors
fROW表示 legend的行数,default 表示是否为默认展示的两种cell info,grp表示是否为分组类型
(2)生成shiny配置文件
1
2
3
4
5
6
7
8
9
|
?makeShinyApp
makeShinyApp(seu, scConf,
gene.mapping = TRUE, # default FALSE, only when ENSEMBL gene id
gex.assay = "RNA", gex.slot = "data",
shiny.title = "scRNA-seq shiny app",
shiny.dir = "shinyApp/")
# 查看结果
list.files("shinyApp")
# [1] "sc1conf.rds" "sc1def.rds" "sc1gene.rds" "sc1gexpr.h5" "sc1meta.rds" "server.R" "ui.R"
|
- 除了其中的
gene.mapping参数(需要在单细胞数据的基因名为ENSEMBL格式时设置为TRUE),其余均为默认参数;
- 此外还有其它参数,包括默认展示的gene,降维类型;
shiny.footnotes参数还支持设置网页的脚注用以交代数据集来源。
(3)最后运行shiny网页即可
1
2
|
# 在本地浏览器的3344端口打开
runApp("shinyApp/", launch.browser = T, port = 3344)
|
3. 模块介绍#
(1)完成上述步骤后即可在本地生成一个shiny网页,下面结合作者分享的示例http://shinycell1.ddnetbio.com/介绍其内容;
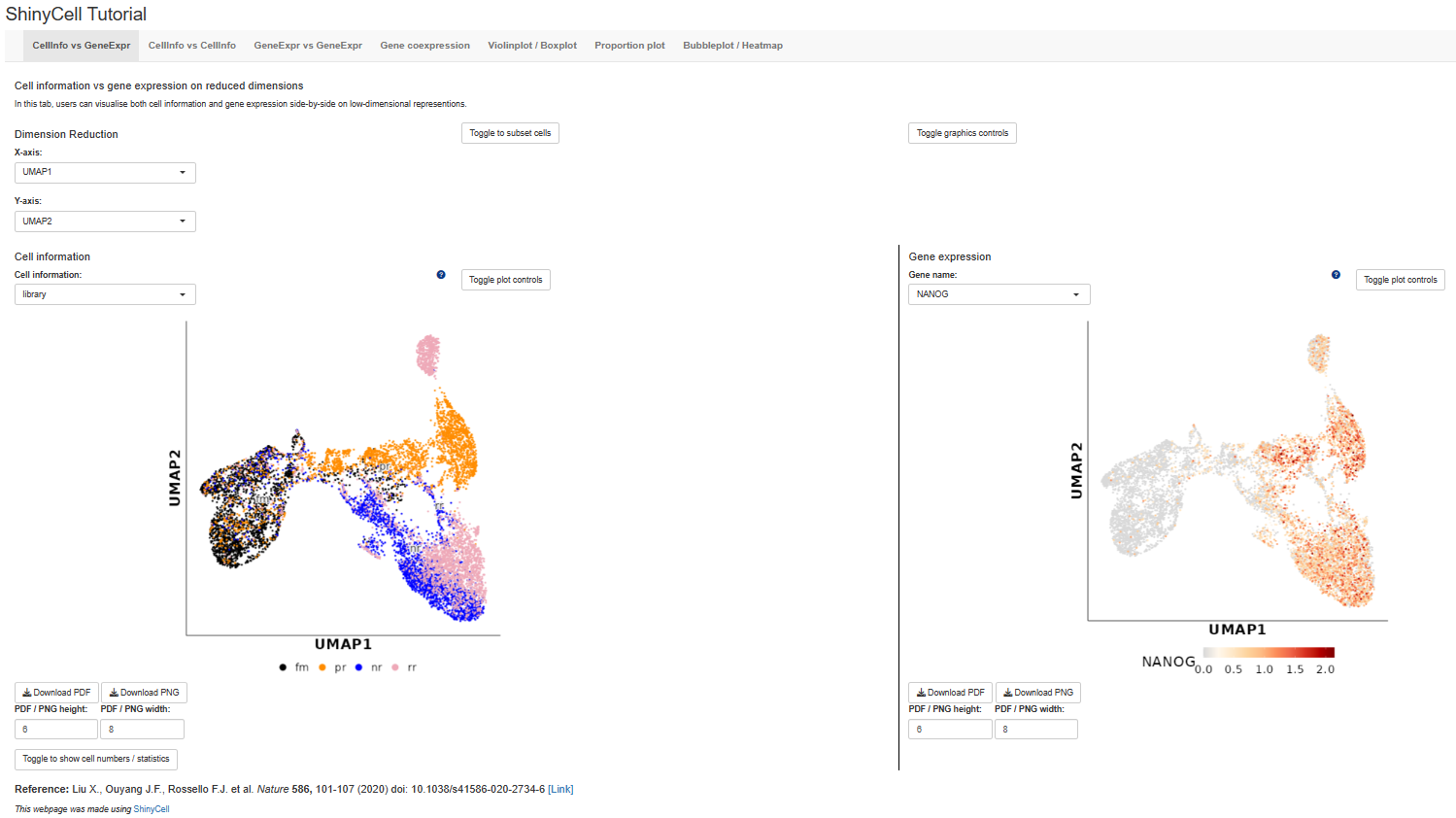
在每个模块中,均有3个Toggle选项支持筛选细胞,调整部分绘图参数等。
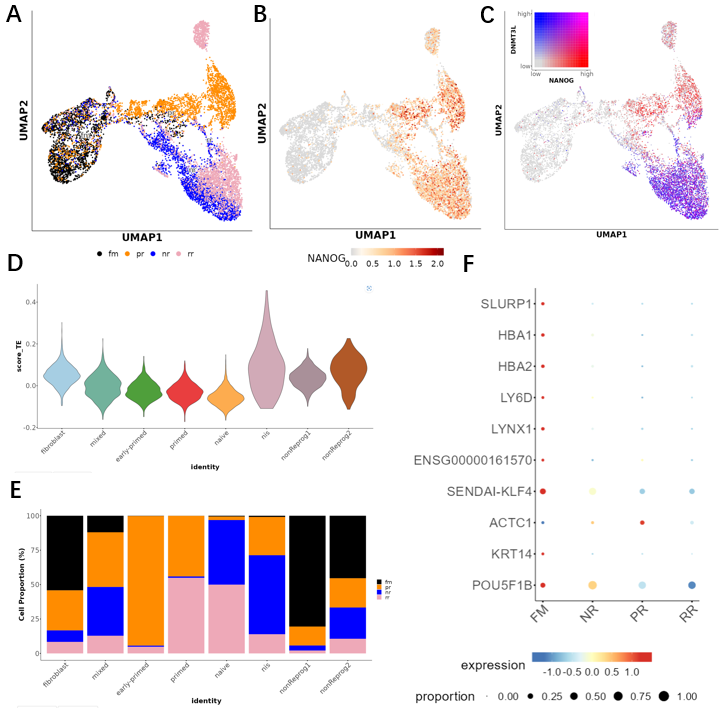
(2)如上图所示,ShinyCell主要支持7个可视化模块;若从绘图类型来说共是6种图
|
模块 |
图形 |
含义 |
e.g. |
| 1 |
CellInfo vs GeneExpr |
降维图 |
对比可视化细胞的分群信息与表达信息 |
A+B |
| 2 |
CellInfo vs CellInfo |
降维图 |
对比可视化细胞的两种不同分群信息 |
A+A |
| 3 |
GeneExpr vs GeneExpr |
降维图 |
对比可视化细胞的两种不同表达信息 |
B+B |
| 4 |
Gene coexpression |
降维图 |
可视化细胞的两种不同表达信息 |
C |
| 5 |
Violinplot / Boxplot |
小提琴图/箱图 |
可视化特定分群下,细胞的表达分布 |
D |
| 6 |
Bubbleplot / Heatmap |
点图/热图 |
可视化特定分群下,细胞的多个表达分布 |
F |
| 7 |
Proportion plot |
柱状图 |
可视化特定分群下,细胞的另一种分群的分布 |
E |
4. 进阶用法#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
|
seu <- readRDS("readySeu_rset.rds")
seu@meta.data = seu@meta.data[,1:6]
scConf = createConfig(seu)
# 展示当前的meta config legend
showLegend(scConf)
# (1)删除若干条细胞注释信息
scConf = delMeta(scConf, c("orig.ident"))
# (2)修改在shiny的细胞注释名title name(fID)
scConf = modMetaName(scConf,
meta.to.mod = c("nUMI", "nGene"),
new.name = c("No. UMIs", "No. detected genes"))
# (3)修改在shiny的细胞注释名level name(fUI)
scConf = modLabels(scConf, meta.to.mod = "library",
new.labels = c("fm", "pr", "nr", "rr"))
# (4)修改在shiny的细胞注释可视化颜色(fCL)
scConf = modColours(scConf, meta.to.mod = "library",
new.colours= RColorBrewer::brewer.pal(n = 4, name = "Set1"))
showLegend(scConf)
# showOrder(scConf)
# scConf = reorderMeta(scConf, rev(scConf$ID))
# showOrder(scConf)
|
如需修改特定meta的level factor顺序,似乎只能在原始单细胞数据的meta中进行修改。
4.2 多个数据集#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
|
## 数据集1
seu <- readRDS("readySeu_rset.rds")
scConf1 = createConfig(seu)
makeShinyFiles(seu, scConf1,
gene.mapping = TRUE,
shiny.prefix = "sc1", # 前缀不同
shiny.dir = "shinyAppMulti/")
## 数据集2
scConf2 = delMeta(scConf1, c("orig.ident"))
makeShinyFiles(seu, scConf2,
gene.mapping = TRUE,
shiny.prefix = "sc2", # 前缀不同
gex.assay = "RNA", gex.slot = "data",
shiny.dir = "shinyAppMulti/")
## 生成shiny配置文件
makeShinyCodesMulti(
shiny.title = "Multi-dataset Tutorial",
shiny.prefix = c("sc1", "sc2"),
shiny.headers = c("Dataset1", "Dataset2"),
shiny.footnotes = "",
shiny.dir = "shinyAppMulti/")
## 运行网页
runApp("shinyAppMulti/", launch.browser = T, port = 3344)
|
4.3 linux平台#
- 对于大型单细胞数据集,需要在服务器linux平台分析时,大体与上述方式类似,简述流程如下。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
# (1) 前期需在linux的R语言环境下安装相关R包,并准备相关数据
# (2) 生成shiny网页文件
seu <- readRDS("readySeu_rset.rds")
scConf = createConfig(seu)
makeShinyApp(seu, scConf, gene.mapping = TRUE,
shiny.title = "ShinyCell Quick Start")
# (3) linux端运行网页,注意需要设置launch.browser参数为FALSE
runApp("shinyApp/", launch.browser = F, port = 3344)
# (4) 本地window端链接:同时win键+R,输入cmd打开命令窗口,根据下述命令进行链接
ssh -L localhost:3344:localhost:3344 username@192.168.212.xxx -p 22
# (5) 最后本地浏览器打开 http://127.0.0.1:3344/ 即可
|