COSG(COSine similarity-based marker Gene identification)是由来自哈佛医学院和Broad研究所博后Ming Dai等人开发,旨在从余弦相似度的角度鉴定cluster的marker gene,于2021年12月被Briefings in Bioinformatics接收。目前已将此方法分别包装为R包与Python包,分别对应Seurat与Scanny分析流程。
1、分析思路#
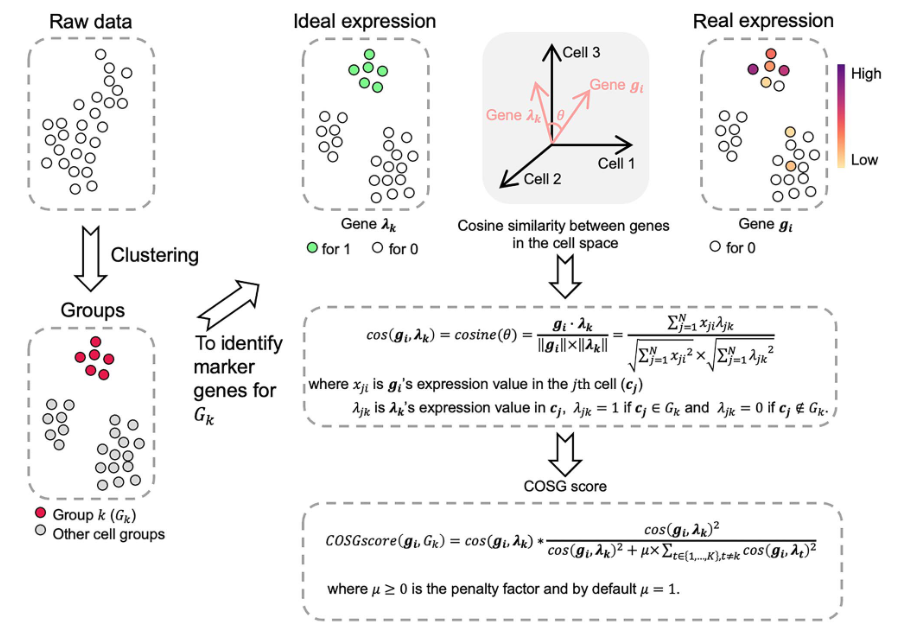
(1)准备好数据, 即标准化后的单细胞表达矩阵,并完成细胞聚类分析(scATAC, ST同理);
(2)对cluster K模拟一个ideal marker gene(仅在k表达,其余cluster不表达);
(3)计算所有真实基因分别与该基因的余弦相似度,并进一步计算出COSG score;
(4)COSG score取值范围为0~1。值越大,表示为cluster specific marker gene;
(5)同理依次计算出其余cluster的marker gene。
2、性能比较#
文章主要与常用的Wilcoxon-test等分析方式在多种数据集上进行比较,概括如下:
(1)在模拟的单细胞数据集中,COSG方法可以最大程度发现每个clutser的marker gene;
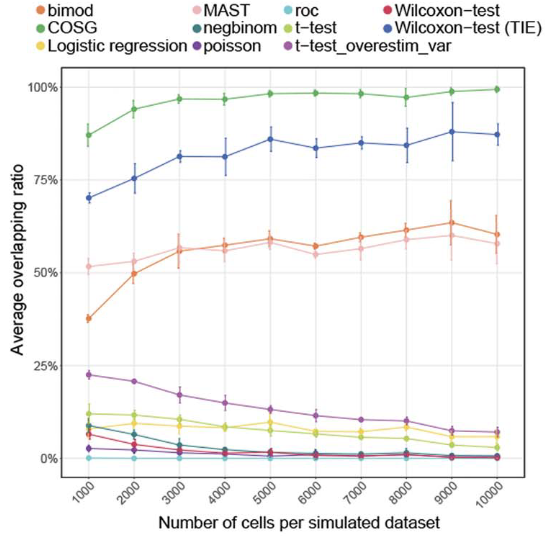
(2)在大规模单细胞数据中, COSG在保持精度的同时分析速度很快;
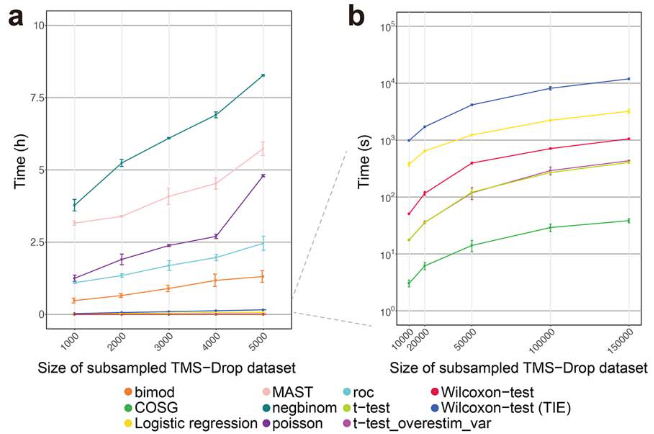
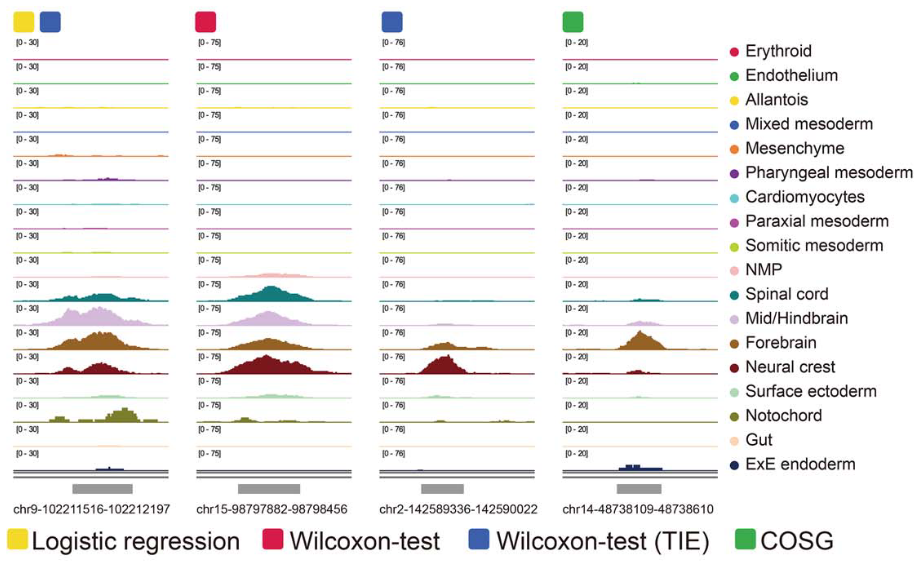
(3)COSG在scATAC以及空间转录组数据上也同样具有较优的表现
3、代码实操#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
remotes::install_github(repo = 'genecell/COSGR')
library(COSG)
library(Seurat)
## 输入数据
data('pbmc_small',package='COSG')
dim(pbmc_small@assays$RNA@data) #标准化矩阵
# [1] 230 80
table(Idents(pbmc_small)) #聚类分群结果
# 0 1 2
# 36 25 19
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
marker_cosg <- cosg(
pbmc_small,
groups='all', #考虑全部分组
assay='RNA',
slot='data',
mu=1, #惩罚项参数,值越大
remove_lowly_expressed=TRUE, #是否过滤低表达基因
expressed_pct=0.1, #设置低表达的阈值
n_genes_user=100 #每个cluster定义Top-N个marker gene
)
head(marker_cosg$names,3)
# 0 1 2
# 1 CD7 S100A8 MS4A1
# 2 CCL5 TYMP CD79A
# 3 GNLY S100A9 TCL1A
head(marker_cosg$scores,3)
# 0 1 2
# 1 0.6391917 0.8954042 0.6922908
# 2 0.6391267 0.8312083 0.5832425
# 3 0.6328148 0.8120045 0.5757478
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
top_list<-c()
for (group in colnames(marker_cosg$names)){
top_i<-marker_cosg$names[group][1:5,1]
top_list<-c(top_list,top_i)
}
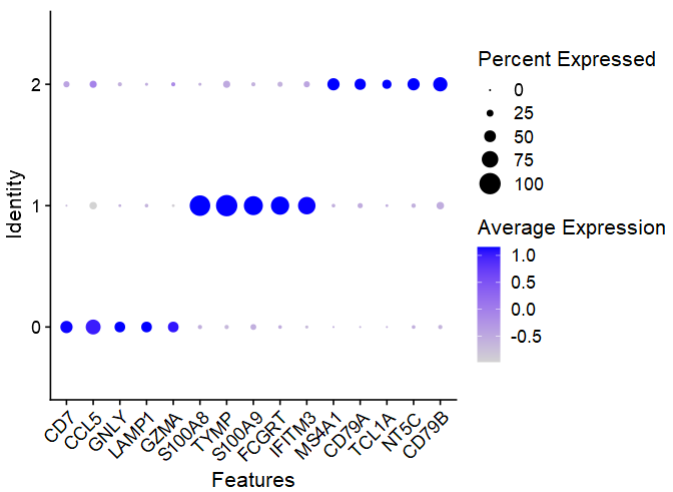
DotPlot(pbmc_small,
assay = 'RNA',
# scale=TRUE,
features = unique(top_list)) + RotatedAxis()
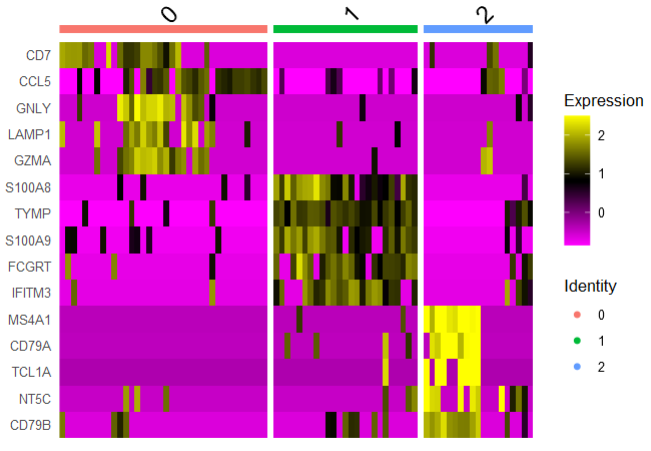
pbmc_small <- ScaleData(pbmc_small, features = top_list)
DoHeatmap(pbmc_small,
assay = 'RNA',
features = top_list)
|