scSTAR是由复旦大学附属中山医院郝洁研究员等人开发的单细胞分析工具包,于2023年2月发表在Briefings in Bioinformatics。该包更加准确地实现同一细胞类型在不同组(正常/疾病)的变化情况下,细胞内部亚型异质性的变化捕捉,在单细胞数据挖掘领域提供新的视角。
- Paper:https://doi.org/10.1093/bib/bbad062
- Github:https://github.com/Hao-Zou-lab/scSTAR
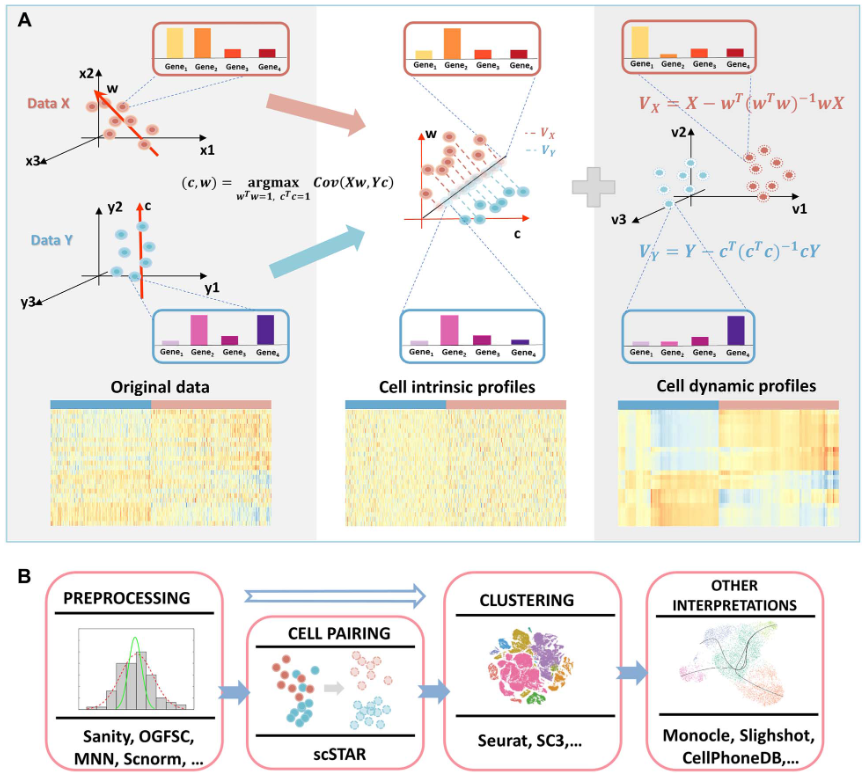
1、文章概述#
示例1:小鼠年老过程中CD4+ T细胞的亚型变化过程
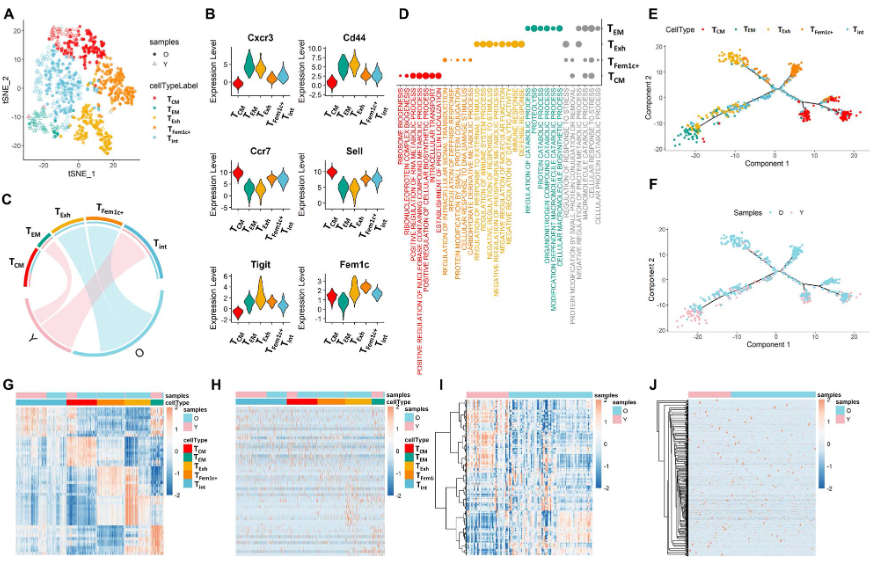
示例2:小鼠年老过程中某一T细胞亚型的比例分布
- activated CD4+ T cells from young and old mice
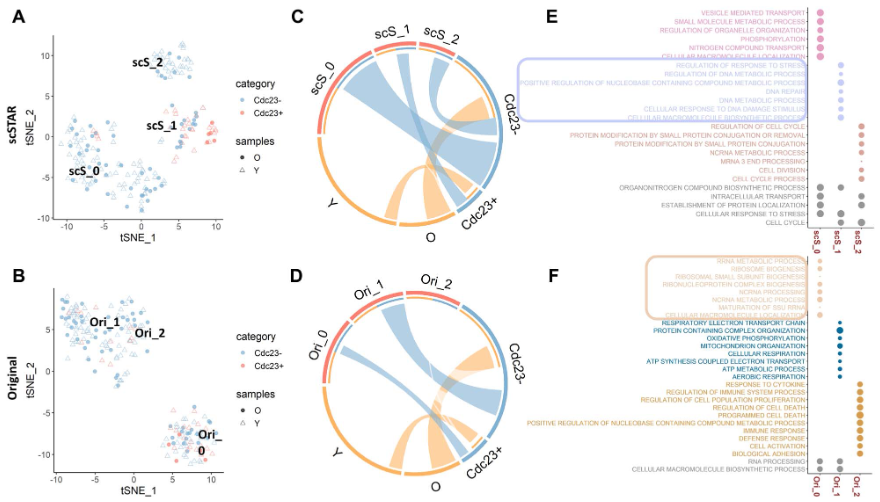
- 根据Cdc23基因是否表达,进一步分为Cdc23+ 与 Cdc23- 两类
- 使用scSTAR分成3个亚群:scS_0/scS_1/scS_2
- scS_1主要包括了Cdc23+,并且主要来自young mice
- scS_2主要包括了Cdc23-,并且主要来自oldmice
- 结论:Cdc23 may play a role in antiageing activities.
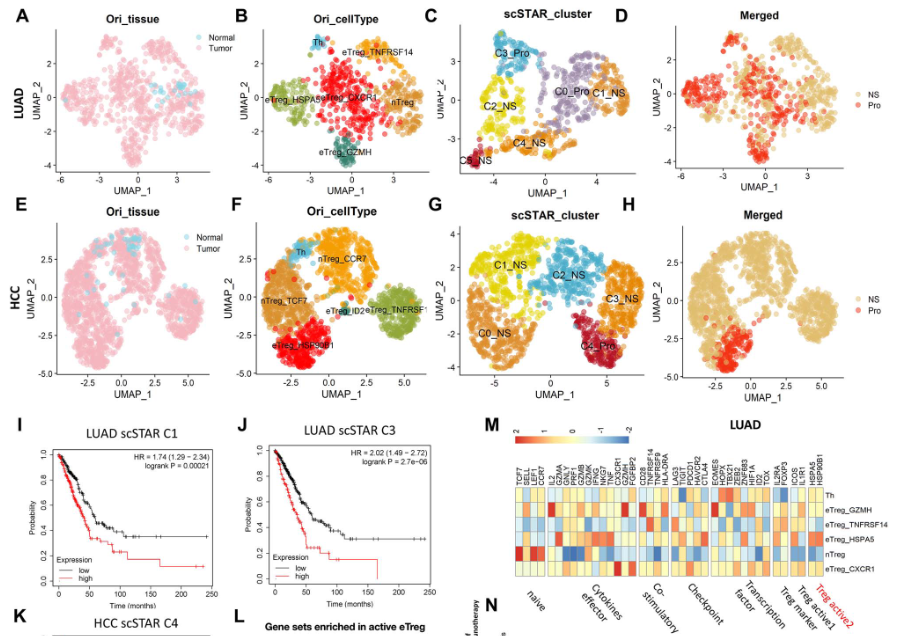
示例3:鉴定出促癌作用的eTreg亚型
- 从正常组织与肺腺癌数据中提取了739个Treg细胞
- 使用scSTAR分为5个亚群:C1-C5
- C1与C3亚群的marker基因与病人生存具有较好的相关性
- 在肝癌数据中得到了相似的发现
- 结论:active Treg subtypes in the tumour microenvironment
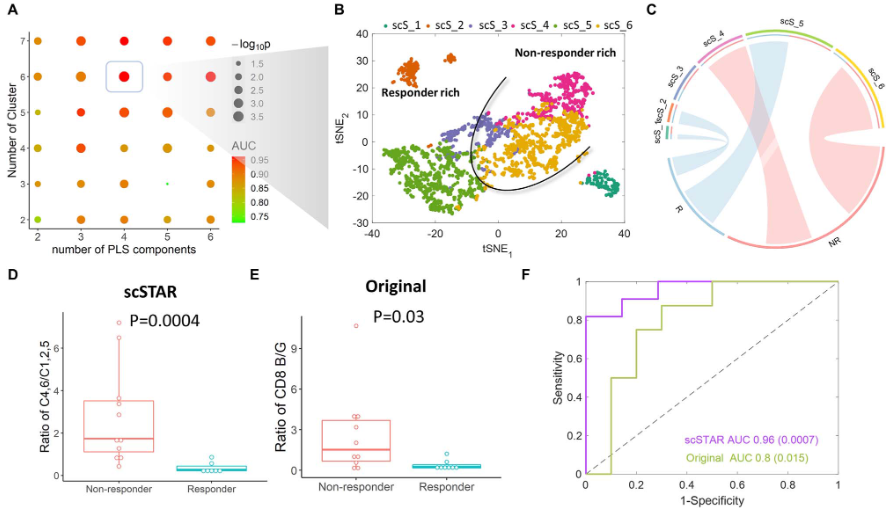
示例4:预测黑色素瘤免疫治疗反应的相关亚型
- 经免疫治疗前后的黑色素瘤样本中的5410个CD8+ T细胞;
- 使用scSTAR将治疗前的CD8+ T分为6个亚群
- 在non-responder组中(C4+C6)/(C1+C2+C5)比值远高于responder组
- 结论:(C4+C6)/(C1+C2+C5)比值对于免疫治疗预后相关
2、R包用法#
本质用法可以理解为对来自两个样本(正常/疾病)的单细胞数据进行“校正批次”
1
2
3
4
5
6
|
library(R.matlab)
library(tsne)
library(pls)
# https://github.com/Hao-Zou-lab/scSTAR/blob/main/scSTAR_0.1.1.0.tar.gz
# install.packages("scSTAR_0.1.1.0.tar.gz", repos = NULL, type="source")
library(scSTAR)
|
- (2)准备log2处理的count矩阵。这里使用示例数据
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
extdir = system.file("extdata",package="scSTAR")
data = readMat(paste0(extdir,'/demo_data.mat'))
geneList = as.matrix(unlist(data$geneList))
data1 = as.matrix((data$data1))
dim(data1)
# [1] 18825 1078
rownames(data1) = geneList
colnames(data1) = paste0("d1_cell",seq(ncol(data1)))
data2 = as.matrix((data$data2))
dim(data2)
# [1] 18825 1388
rownames(data2) = geneList
colnames(data2) = paste0("d2_cell",seq(ncol(data2)))
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
## step-1: 挑选基因
data = cbind(data1, data2)
idx_OGFSC = filter_gene(data, nBins = 20, plot_option = 1)$OGFSC_idx
length(idx_OGFSC)
# [1] 6683
head(idx_OGFSC)
# [1] 5 6 10 11 12 17
Ctr_filtered = data1[idx_OGFSC,]
Case_filtered = data2[idx_OGFSC,]
## step-2: 鉴定细胞并"校正"
anchorCells = findAnchors(Ctr_filtered, Case_filtered, 3)
# When PLScomp = 0, the number of PLScomp can be automatically estimated.
# suggest 3-5
out = scSTAR(Ctr_filtered, Case_filtered, anchorCells, PLScomp = 4)
names(out)
# [1] "Case_kinetics" "idx_DE_up" "Ctr_corrected" "Case_corrected" "Ctr_kinetics" "idx_DE_down"
# 查看函数帮助文档,了解每个的含义
|

1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
data2 = cbind(out$Case_kinetics, out$Ctr_kinetics)
rownames(data2) = geneList[idx_OGFSC]
dim(data2)
# [1] 6683 2466
library(Seurat)
library(tidyverse)
sce = Seurat::CreateSeuratObject(data2)
sce@assays$RNA@data = data2
sce = sce %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA() %>%
RunUMAP(dims = 1:30)
# DimPlot(sce, reduction = "umap")
sce = sce %>%
FindNeighbors(dims = 1:30) %>%
FindClusters(resolution = c(0.01, 0.05, 0.1))
diff_wilcox = FindAllMarkers(sce, assay = "RNA", slot = "data")
|