来自密歇根大学医学院的计算医学与生物信息学部的LanaX.Garmire(拉娜·加米尔)团队近日于Nature Communication发表一篇基于单细胞转录组数据药物重定向研究的研究,并开发了相关R包。如下简单学习下该方法的原理以及R包使用方法。
- Paper:ASGARD is A Single-cell Guided Pipeline to Aid Repurposing of Drugs
- Date:10 February 2023
- DOI:https://doi.org/10.1038/s41467-023-36637-3
- Github:https://github.com/lanagarmire/Asgard
1、算法简介#
(1)基本的计算思路与CMAP方法类似,即疾病的差异基因方向与药物干扰细胞系方向相反;
(2)使用对照组与疾病组单细胞数据分析每个细胞亚群的差异基因及其候选药物列表;
(3)合并不同细胞群的结果,得到单药物对于所有细胞群的综合打分,以评价其重定向的潜力。
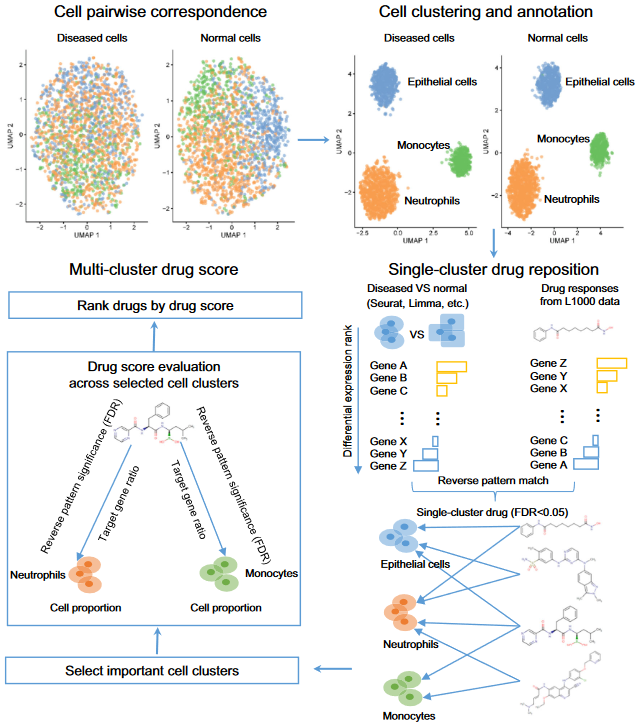
(4)单药-单细胞群的打分
- 预先将疾病细胞群差异基因由低到高排序,而药物干扰基因由高到低排序。如果排序完全相反则认为药物越好;
- 该包采用了β分布,计算了同一基因的在疾病/药物的排名最小值/最大值的出现的概率,进而求出P值(Min-P);
- 如果P值小的基因越多,表明基因在疾病与药物的排名一致性越好。
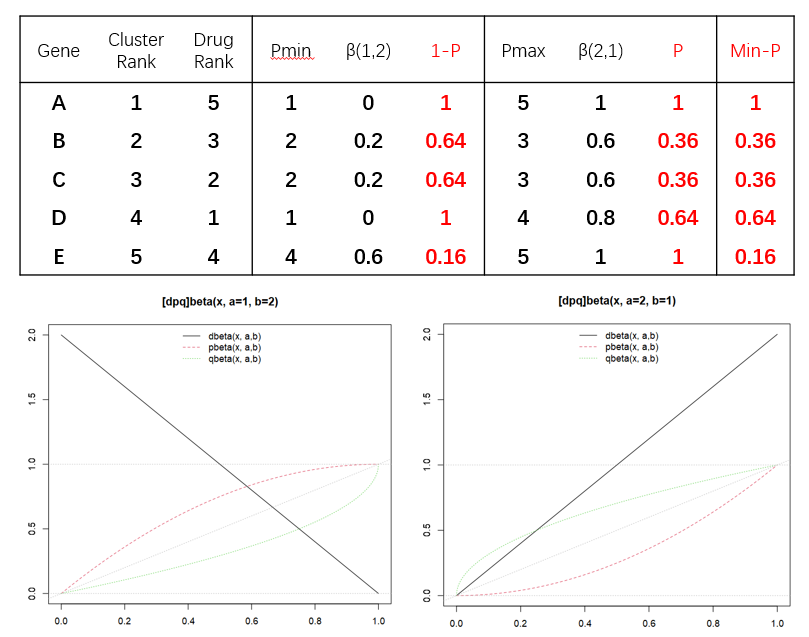
- 设定一定的阈值(默认为0.05)计算所有药物实验结果P值显著的基因数;
- 由于一个药物可因浓度剂量等涉及多种实验,所以再进一步根据KS统计量计算单药的P值,作为单药对于单细胞群的打分。
(5)单药-多细胞群打分。
- 按如下公式计算综合得分,其中考虑3项因素:细胞群比例, 单药对于细胞群的P值,药物可逆转的细胞群差异基因比例。
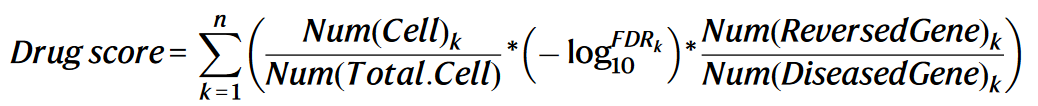
- 基于单药对于不同细胞群的P值计算出fisher-based combined P值作为综合得分的P值。
2、文章概述#
实验数据集主要包括3个(1)乳腺癌(2)白血病(3)COVID-19
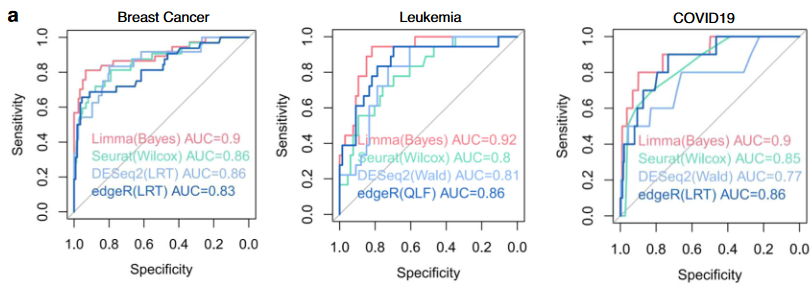
(1) 差异基因分析方法:文章首先测试对于不同数据哪一种差异基因分析工具最终计算得到的AUC指标最高(FDA或临床报道作为阳性药)
-
Limma:Bayes,trend,voom
-
Seurat:Wilcox,t-test,LR
-
DEseq2:Wald,LRT
-
edgeR:LRT,QLF
如下图发现,limma-bayes方法结果针对3个数据集均较优,后面的差异分析将使用这种分析方法。
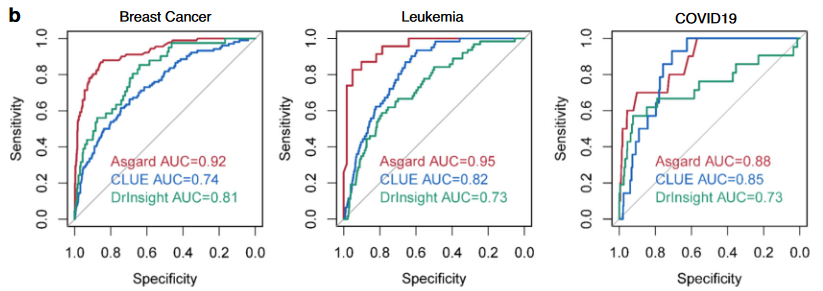
(2)对比基于Bulk RNA-seq数据的重定向方法(CLUE,DrInsight),将单细胞数据转换成pseudobulk RNA-Seq data。
如下图发现ASGARD在三个数据集中均表现最优。
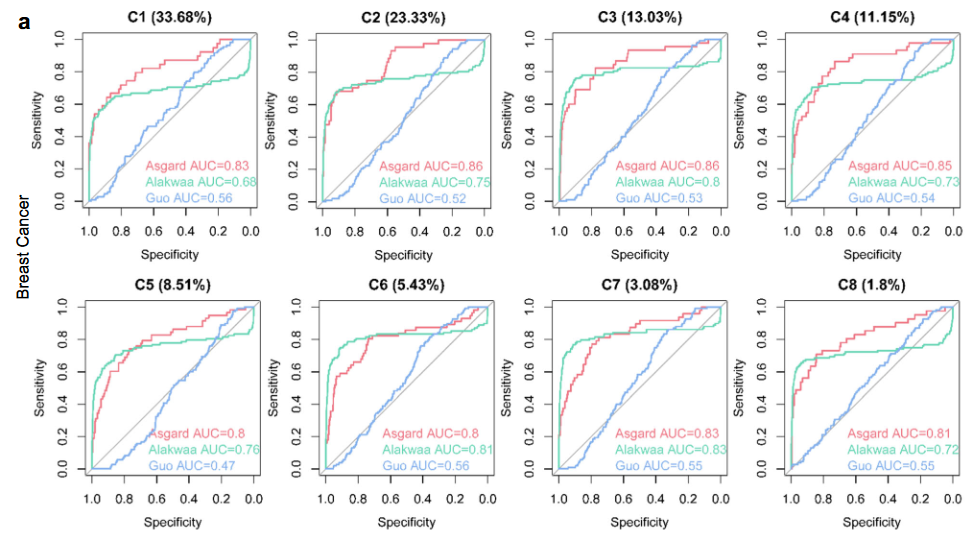
(3)对比另外两种同样基于单细胞数据的重定向方法,由于后两者只能针对单独cluster进行预测,所以仅使用ASGARD的cluster预测模式。尽管如此,仍然发现ASGARD在3种数据集中均表现最优。
(4)之后文章就ASGARD对于上述3种数据集的预测Top药物结合文献、靶点通路等角度说明预测结果的可靠性;具体可参考原文。
3、R包用法#
1
2
3
|
library(tidyverse)
library(Seurat)
library(Asgard)
|
3.1 准备数据#
- 药物数据:下载CMAP数据后,然后按细胞系所属的组织器官系统拆分成若干类
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
## linux下载
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_cell_info_2017-04-28.txt.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_sig_info_2017-03-06.txt.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_gene_info_2017-03-06.txt.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_cell_info.txt.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx.gz
# wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_sig_info.txt.gz
## 拆分数据,只需完成一次即可(已上传至网盘)
# PrepareReference(cell.info="GSE70138_Broad_LINCS_cell_info_2017-04-28.txt",
# gene.info="GSE70138_Broad_LINCS_gene_info_2017-03-06.txt",
# GSE70138.sig.info = "GSE70138_Broad_LINCS_sig_info_2017-03-06.txt",
# GSE92742.sig.info = "GSE92742_Broad_LINCS_sig_info.txt",
# GSE70138.gctx = "GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx",
# GSE92742.gctx = "GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx",
# Output.Dir = "DrugReference/"
# )
|
- 疾病数据:github给了原文分析所用的乳腺癌数据,这里简单以PBMC3K模拟一下
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
|
#devtools::install_github('satijalab/seurat-data')
SeuratData::InstallData("pbmc3k")
data("pbmc3k")
sce = pbmc3k
sce$celltype = as.character(sce$seurat_annotations)
# 以其中4种T细胞亚类为例
sce = sce[, str_detect(sce$celltype,"CD")]
set.seed(42)
sce$sample <- sample(c("before", "after"), size = ncol(sce),
replace = TRUE)
table(sce$celltype, sce$sample)
# after before
# CD14+ Mono 236 244
# CD8 T 133 138
# Memory CD4 T 234 249
# Naive CD4 T 358 339
Case=c("before")
Control=c("after")
|
3.2 细胞亚群DEG#
- 该包Github首页教程中给出了全部4种差异分析的方法,这里以Seurat包的默认方法为例
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
DefaultAssay(sce) <- "RNA"
Gene.list <- list()
C_names <- NULL
for(i in unique(sce@meta.data$celltype)){
# i = unique(sce@meta.data$celltype)[1]
Idents(sce) <- "celltype"
c_cells <- subset(sce, celltype == i)
Idents(c_cells) <- "sample"
C_data <- FindMarkers(c_cells, ident.1 = Case, ident.2 = Control)
C_data_for_drug <- data.frame(row.names=row.names(C_data),score=C_data$avg_log2FC,adj.P.Val=C_data$p_val_adj,P.Value=C_data$p_val) ##for Seurat version > 4.0, please use avg_log2FC instead of avg_logFC
Gene.list[[i]] <- C_data_for_drug
C_names <- c(C_names,i)
}
names(Gene.list) <- C_names
lapply(Gene.list, head)
|
3.3 药物干扰DEG#
1
2
3
4
5
6
7
|
my_gene_info<-read.table(file="DrugReference/breast_gene_info.txt",sep="\t",header = T,quote = "")
my_drug_info<-read.table(file="DrugReference/breast_drug_info.txt",sep="\t",header = T,quote = "")
drug.ref.profiles = GetDrugRef(drug.response.path = 'DrugReference/breast_rankMatrix.txt',
probe.to.genes = my_gene_info,
drug.info = my_drug_info)
# 包含两个元素的列表:[1] 药物的meta信息 [2] 药物的干扰基因排名结果
# 值得注意的是每个药物会因不同浓度,不同剂量等有不同的实验结果
|
3.4 细胞群药物#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
Drug.ident.res = GetDrug(gene.data = Gene.list,
CEG.threshold = 0.05, #判定每个基因是否显著的阈值
drug.ref.profiles = drug.ref.profiles,
repurposing.unit = "drug",
connectivity = "negative", #从小到大开始排序
drug.type = "FDA") #c("FDA","compounds","all")
head(Drug.ident.res[[1]])
# Drug.name Drug.id P.value FDR
# 1 imatinib BRD-K92723993 1.491597e-07 0.0001971891
# 2 palbociclib BRD-K51313569 5.449091e-05 0.0360184923
# 3 auranofin BRD-A79465854 5.385982e-04 0.2373422941
# 4 nilotinib BRD-K81528515 8.217091e-04 0.2715748734
# 5 mebendazole BRD-K77987382 2.515145e-03 0.6650042235
# 6 guaifenesin BRD-A90515964 3.726919e-03 0.8211644934
str(Asgard::FDA.drug)
# chr [1:4014] "lepirudin" "cetuximab" "dornase alfa" "denileukin diftitox" ...
|
3.5 药物综合分数#
- 由于随机模拟数据的原因,且算法(已经向GITHUB issue)的问题,如下结果的Drug.therapeutic.score均为NAN。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
GSE92742.gctx.path="GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx"
GSE70138.gctx.path="GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx"
Tissue="breast"
Drug.score<-DrugScore(SC.integrated=sce,
Gene.data=Gene.list,
Cell.type=NULL,
Drug.data=Drug.ident.res,
FDA.drug.only=TRUE,
Case=Case,
Tissue=Tissue,
GSE92742.gctx=GSE92742.gctx.path,
GSE70138.gctx=GSE70138.gctx.path)
head(Drug.score)
# Drug.therapeutic.score P.value FDR
# abiraterone NaN 0.1164141 0.999979
# acamprosate NaN 0.3386581 0.999979
# acarbose NaN 0.4719701 0.999979
|