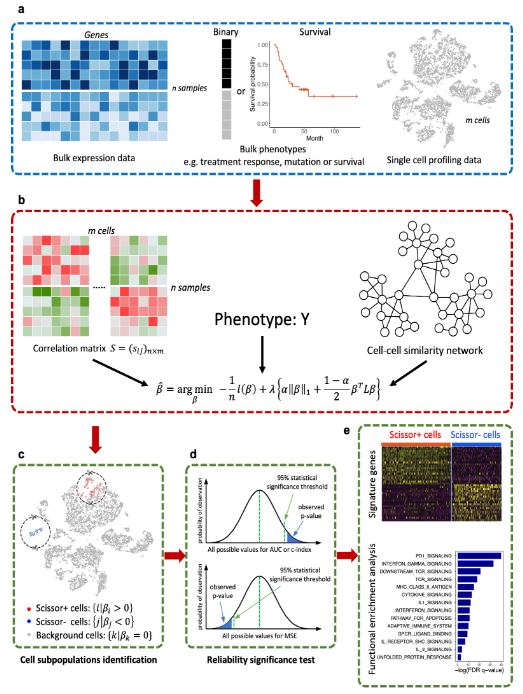
Scissor是2022年发表在Nature Biotechnology的一个单细胞数据分析工具包。该包通过纳入Bulk表达与表型数据,试图分析与样本表型(正/负)高度相关的单细胞亚群。
- 原始论文:https://doi.org/10.1038/s41587-021-01091-3
- 官方手册:https://github.com/sunduanchen/Scissor
- R包安装
1
|
devtools::install_github('sunduanchen/Scissor')
|
- 简介:该包首先基于表达数据计算每个单细胞与bulk样本的相关性,筛选相关性较好的细胞群;然后进一步结合表型信息,通过回归模型并加上惩罚项选出最相关的亚群。之后基于两类亚群(正/负相关)可通过差异分析、通路分析、signature分析以及单细胞相关高级分析对该亚群进行多角度的个性分析。具体可参考原论文的分析示例。
0、示例数据#
参考官方手册,使用肺腺癌(LUAD)示例数据如下:
1
2
3
4
5
6
7
|
# https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/scRNA-seq.RData
load("./scRNA-seq.RData")
dim(sc_dataset)
# [1] 33694 4102
#上述为matrix格式,需要转为seurat对象并完成预处理
?Seurat_preprocessing
sc_dataset = Seurat_preprocessing(sc_dataset, verbose = F)
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
##表达矩阵
# https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/TCGA_LUAD_exp1.RData
load("./TCGA_LUAD_exp1.RData")
dim(bulk_dataset)
# [1] 56716 471
##表型信息
# https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/TCGA_LUAD_survival.RData
load("./TCGA_LUAD_survival.RData")
all(colnames(bulk_dataset) == bulk_survival$TCGA_patient_barcode)
# [1] TRUE
phenotype <- bulk_survival[,2:3]
colnames(phenotype) <- c("time", "status")
head(phenotype)
# time status
# 1 1158 0
# 2 121 1
# 3 607 0
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
## 表达矩阵
# https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/TCGA_LUAD_exp2.RData
load("./TCGA_LUAD_exp2.RData")
dim(bulk_dataset)
# [1] 56716 498
## 表型信息
# https://xialab.s3-us-west-2.amazonaws.com/Duanchen/Scissor_data/TCGA_LUAD_TP53_mutation.RData
load("./TCGA_LUAD_TP53_mutation.RData")
table(TP53_mutation)
# 0 1
# 243 255
phenotype <- TP53_mutation
tag <- c('wild-type', 'TP53 mutant')
|
1、函数用法#
1
2
|
library(Scissor)
?Scissor()
|
Scissor()是该包的核心分析函数,相关参数与注意事项如下:
1.1 输入数据#
首先依次输入3类数据:
- (1)bulk_dataset :Bulk多样本表达矩阵;
- (2)sc_dataset :sc_dataset单细胞表达数据,建议使用预处理的Seurat对象;
- (3)phenotype : Bulk多样本表型数据。
1.2 模型参数#
- (1)alpha :表示对回归模型的惩罚项;值越大,最终选择的细胞数越少。
- (2)cutoff :表示预期最大的选择细胞数比例,用以指导优化alpha参数
- (3)family : 表示回归模型的类型。gaussian-线性回归;binomial-二分类;cox-生存预后
默认alpha=NULL, cutoff=0.2,训练模型时从小到大遍历alpha值,直至达到cutoff所设的阈值。
此外也可以指定alpha为一个或多个候选值,进行遍历。
1.3 临时文件#
- (1)Save_file :用以保存训练模型所需的中间数据,节省第二次训练的时间;包括如下5项
- Expression_bulk - bulk表达矩阵
- Expression_cell - 单细胞表达矩阵
- network - 单细胞相似度网络
- X - 每个细胞与每个bulk样本的相关性网络
- Y - bulk表型数据
- (2)Load_file :第二次训练时直接加载上次保存的中间数据,用以节省时间
2、生存相关亚群#
1
2
3
4
5
6
7
8
9
10
11
12
|
infos1 = Scissor(bulk_dataset, sc_dataset, phenotype, family = "cox",
alpha = NULL, cutoff = 0.2,
Save_file = 'Scissor_LUAD_survival.RData')
str(infos1)
# List of 4
# $ para :List of 3
# ..$ alpha : num 0.01
# ..$ lambda: num 0.156
# ..$ family: chr "cox"
# $ Coefs : num [1:4102] 0 0 0 0 0 ...
# $ Scissor_pos: chr [1:573] "AAACGGGAGTCCATAC_19" "AAACGGGTCACATACG_19" "AAAGTAGAGGAGCGAG_19" "AAAGTAGCAACTGCTA_18" ...
# $ Scissor_neg: chr [1:20] "AACGTTGCAAGCCCAC_16" "AACTCTTTCGAACTGT_20" "ACGAGCCCAACACCTA_20" "ACGCCAGTCCTCCTAG_20" ...
|
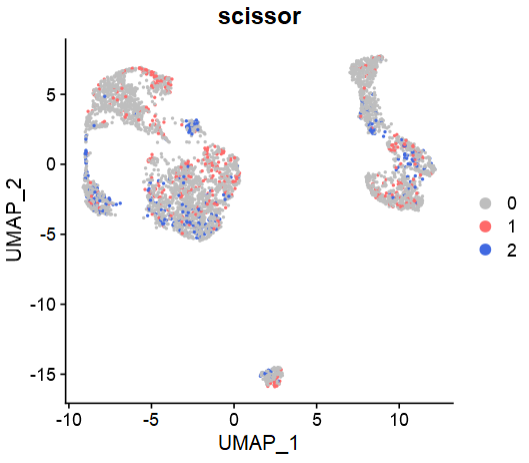
- 如上,Scissor分析结果包含4项内容:(1)训练参数;(2)每个细胞与bulk样本的回归系数;(3)**Scissor+**细胞标签;(4)**Scissor-**细胞标签;
- 对于Scissor+与Scissor-两类亚群,需结合表型信息进行解读。在这里:Scissor+表示与预后较差表型相关;Scissor-则与之相反。
1
2
3
4
5
6
7
8
9
|
Scissor_select <- rep(0, ncol(sc_dataset))
names(Scissor_select) <- colnames(sc_dataset)
Scissor_select[infos1$Scissor_pos] <- 1
Scissor_select[infos1$Scissor_neg] <- 2
sc_dataset <- AddMetaData(sc_dataset, metadata = Scissor_select, col.name = "scissor")
# 0 1 2
# 3509 573 20
DimPlot(sc_dataset, reduction = 'umap', group.by = 'scissor',
cols = c('grey','indianred1','royalblue'))
|
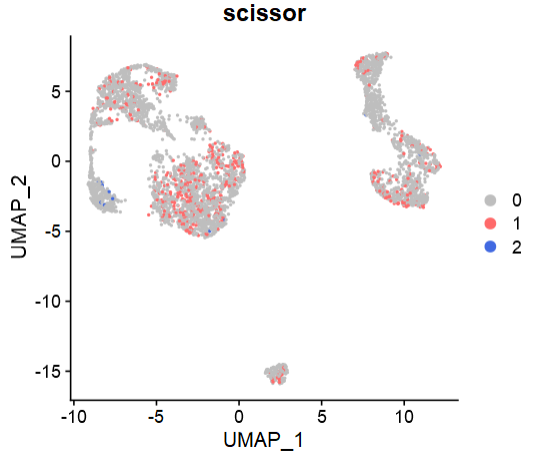
3、TP53相关亚群#
1
2
3
4
5
6
7
8
9
10
11
12
13
|
infos2 <- Scissor(bulk_dataset, sc_dataset, phenotype, tag = tag,
family = "binomial",
alpha = NULL, cutoff = 0.2,
Save_file = "Scissor_LUAD_TP53_mutation.RData")
str(infos2)
Scissor_select <- rep(0, ncol(sc_dataset))
names(Scissor_select) <- colnames(sc_dataset)
Scissor_select[infos2$Scissor_pos] <- 1
Scissor_select[infos2$Scissor_neg] <- 2
sc_dataset <- AddMetaData(sc_dataset, metadata = Scissor_select, col.name = "scissor")
DimPlot(sc_dataset, reduction = 'umap', group.by = 'scissor',
cols = c('grey','indianred1','royalblue'))
|