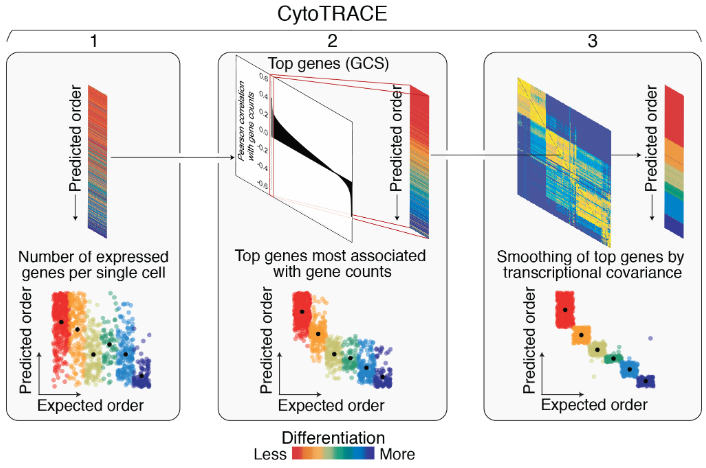
CytoTRACE是根据单细胞表达矩阵推断细胞分化轨迹的工具,目前提供网页端与R包两种形式。如下。简单学习其R包用法。
1、安装R包#
根据官方手册,需要手动R包文件,自行安装。如下笔记在Linux的R环境中进行学习。
- R包下载:https://cytotrace.stanford.edu/CytoTRACE_0.3.3.tar.gz
1
|
devtools::install_local("CytoTRACE_0.3.3.tar.gz")
|
如上,首先会自动安装若干依赖R包。如果未能成功安装,可单独使用conda安装,例如sva等
1
2
|
conda install -c bioconda bioconductor-sva
conda install -c conda-forge r-ncdf4
|
- 安装python模块:针对多样本的轨迹分析函数需要用到两个python模块,需要安装
1
2
|
pip install scanoramaCT
pip install numpy
|
在如上的scanoramaCT安装过程中也会下载若干依赖包。若安装失败,同样可单独conda安装。例如:
1
|
conda install -c conda-forge python-annoy
|
2、CytoTRACE分析#
CytoTRACE包架构很简单,包含两个分析函数、两个绘图函数,以及其余的4个示例数据。
-
(1)输入数据
- 原始单细胞count表达矩阵:行名为基因名,列名为细胞名的dataframe/matrix;
- 细胞类型标签:字符串格式。值表示注释的细胞类型;名字对应表达矩阵的行名。
- 降维坐标(optional):dataframe格式,包含两列。行名对应表达矩阵的列名。
-
(2)CytoTRACE()单样本分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
dim(marrow_10x_expr)
# [1] 13526 3427
length(marrow_10x_pheno)
# [1] 3427
head(table(marrow_10x_pheno))
# Erythrocytes Erythroid progenitors and erythroblasts
# 142 268
# Granulocyte progenitors Granulocytes
# 330 770
# Immature B cells Macrophages
# 299 222
## 多线程分析
results = CytoTRACE(marrow_10x_expr,
ncores = 8, subsamplesize = 1000)
length(results) # 8
|
如上,分析结果是长度为8的列表格式,其中比较重要的前三项。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
## (1) 第一项表示:每个细胞的轨迹分数,取值范围在0~1。越接近0,分化潜能越大;反之越小。
head(results[[1]])
# 10X_P7_2_AAACCTGCAGTAACGG 10X_P7_2_AAACGGGAGGACGAAA 10X_P7_2_AAACGGGAGGTACTCT
# 0.62346760 0.35901926 0.73555166
# 10X_P7_2_AAACGGGAGGTGCTTT 10X_P7_2_AAACGGGAGTCGAGTG 10X_P7_2_AAAGATGAGCTTCGCG
# 0.09457093 0.23467601 0.42644483
## (2) 第二项表示:每个细胞的分化潜能的排名
# 10X_P7_2_AAACCTGCAGTAACGG 10X_P7_2_AAACGGGAGGACGAAA 10X_P7_2_AAACGGGAGGTACTCT
# 713 411 841
# 10X_P7_2_AAACGGGAGGTGCTTT 10X_P7_2_AAACGGGAGTCGAGTG 10X_P7_2_AAAGATGAGCTTCGCG
# 109 269 488
## (3) 第三项表示:每个基因表达与细胞群分化轨迹相关性
head(results[[3]])
# Rpl4 Eef1a1 Rps5 Rps3a Rpl13a Rps4x
# 0.9234229 0.9225254 0.9222174 0.9216470 0.9196403 0.9161128
## 具体每项意义可通过函数帮助文档获取
?CytoTRACE
|
1
2
3
4
5
6
|
datasets <- list(marrow_10x_expr, marrow_plate_expr)
multi_results <- iCytoTRACE(datasets,
ncores = 8, subsamplesize = 1000)
## 分析结果同样是长度为8的列表格式
## 具体每项意义可通过函数帮助文档获取
?iCytoTRACE
|
3、结果可视化#
以上述的CytoTRACE()函数分析结果results为例
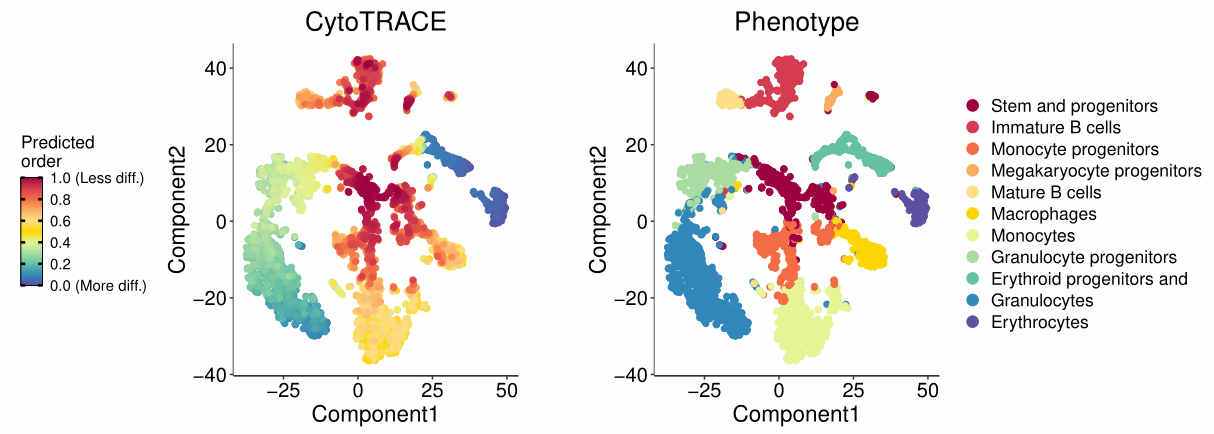
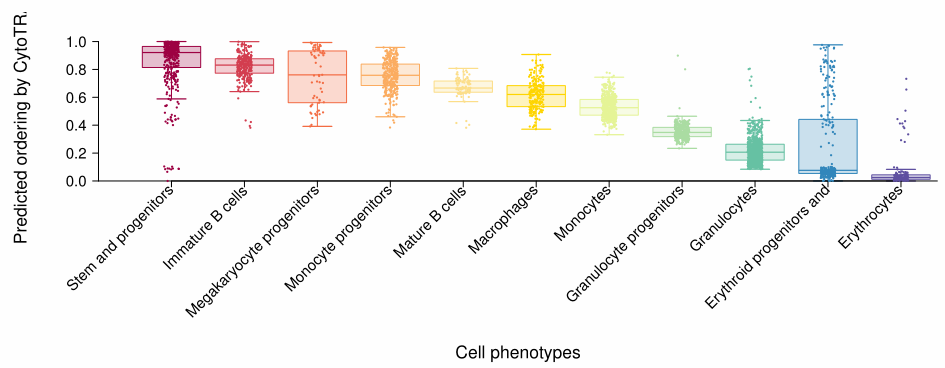
- (1)plotCytoTRACE
- 函数会自动绘制两个图形,并保存在特定路径;
- 如未提供embedding,函数自动采用tsne降维。
1
2
3
4
5
6
7
|
plotCytoTRACE(
cyto_obj = results,
phenotype = marrow_10x_pheno, #细胞类型注释
# gene = "Kit", #是否映射特定基因表达
emb = NULL, #是否提供细胞降维坐标
outputDir = "./" #图片储存路径
)
|
1
2
3
4
5
6
|
plotCytoGenes(
cyto_obj = results,
numOfGenes = 10,
colors = c("darkred", "navyblue"),
outputDir = "./"
)
|
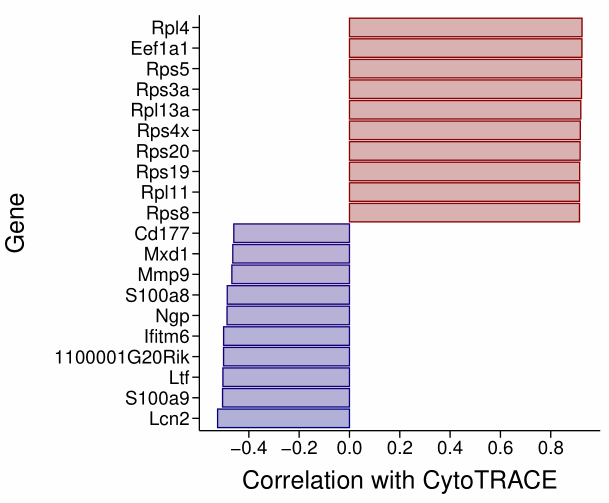
如上图形均可根据分析结果数据自行绘制,或者添加到Seura对象的meta.data中进行可视化。