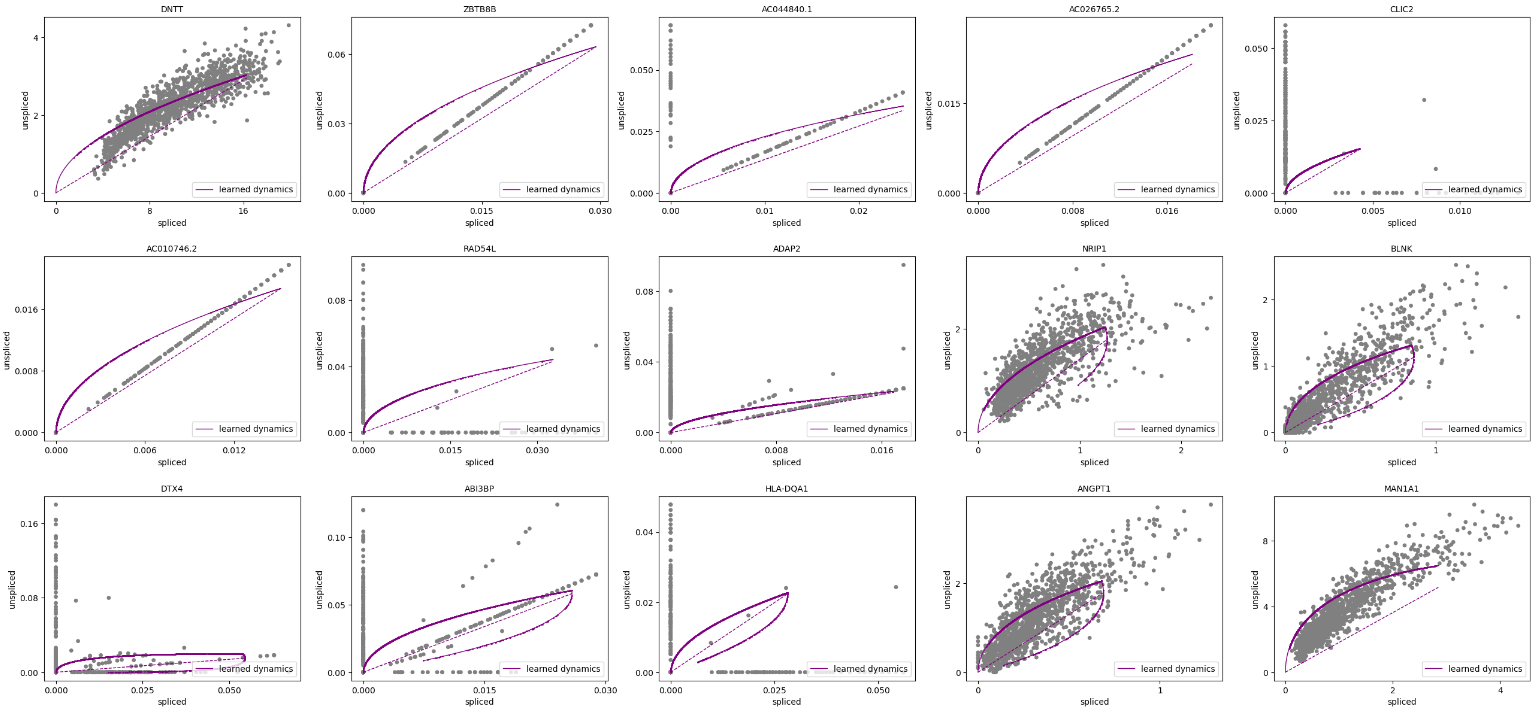
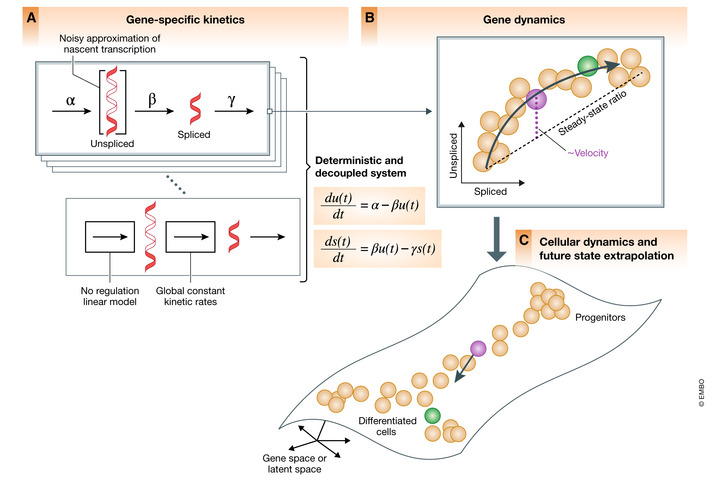
RNA velocity(RNA速率)分析是基于单细胞转录组测序数据分析细胞发育状态动力学的方法。简单来说,该模型根据测序read片段属于unspliced pre-mrna以及spliced mature mrna的比例推测单细胞的发育轨迹。 高比例的unspliced pre-mrna的占比越高,velocity速率越大,表明在之后阶段中将产生高表达趋势。
如下将主要学习分析流程,算法原理可参考原始论文:
- 2018 | Natue | DOI: 10.1038/s41586-018-0414-6
- 2020 | Nature Biotechnology |DOI: 10.1038/s41587-020-0591-3
分析流程参考教程:
- 上游比对:https://velocyto.org/velocyto.py/index.html
- 下游分析:https://smorabit.github.io/tutorials/8_velocyto/
1、上游比对
- 单细胞测序技术有很多,如下以常用的10X genomics的测序结果为例。
- 首先根据原始测序结果fastq.gz,经10X官方的cellranger软件进行比对(使用方法参考之前笔记)
- 根据
filtered_feature_bc_matrix文件夹结果使用Seurat包进行单细胞常规分析; - 根据比对的bam文件,使用velocyto软件分别识别出比对到unspliced与spliced mRNA。
- 根据
示例数据是GSE178911中的GSM5400792样本,为人体急性髓系白血病(AML)骨髓细胞。已经过cellranger比对,结果保存在
201008_D0_scRNA_fastq文件夹。
1 2 3 4 5 6 7 8 9 10 11 12 13tree -L 1 ./201008_D0_scRNA_fastq/outs/ # ./201008_D0_scRNA_fastq/outs/ # ├── cellsorted_possorted_genome_bam.bam # ├── filtered_feature_bc_matrix # ├── filtered_feature_bc_matrix.h5 # ├── filtered_feature_bc_matrix.tar.gz # ├── metrics_summary.csv # ├── molecule_info.h5 # ├── possorted_genome_bam.bam # ├── possorted_genome_bam.bam.bai # ├── raw_feature_bc_matrix # ├── raw_feature_bc_matrix.h5 # └── web_summary.html
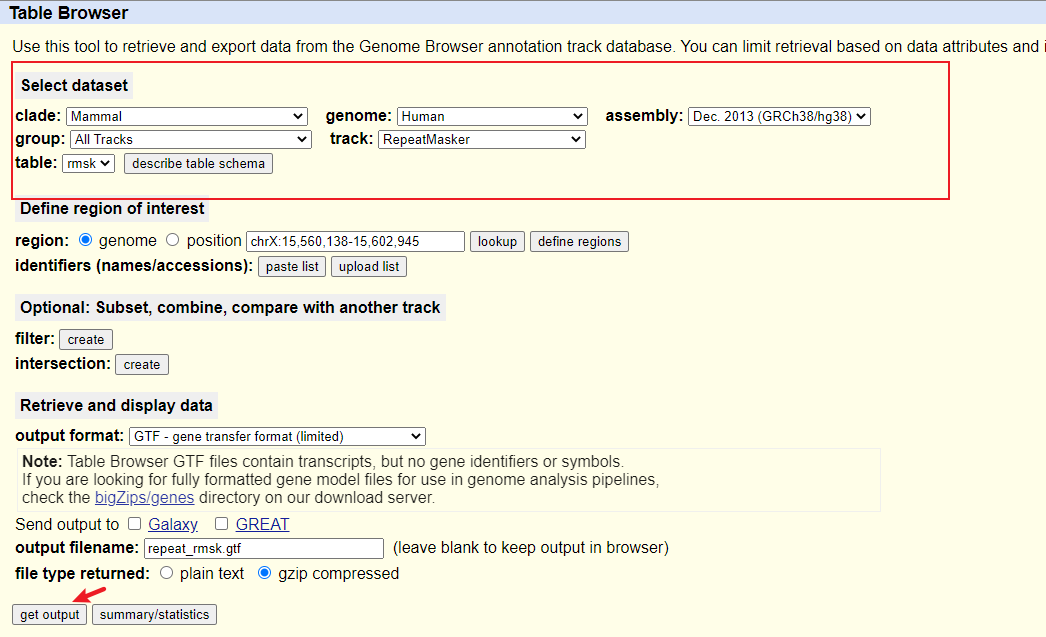
1.1 velocyto分析
|
|
rmsk_gtf下载链接:https://genome.ucsc.edu/cgi-bin/hgTables?hgsid=611454127_NtvlaW6xBSIRYJEBI0iRDEWisITa&clade=mammal&org=Human&db=0&hgta_group=allTracks&hgta_track=rmsk&hgta_table=rmsk&hgta_regionType=genome&position=&hgta_outputType=gff&hgta_outFileName=mm10_rmsk.gtf
|
|
如上,每个样本都会产生一个loom文件结果。
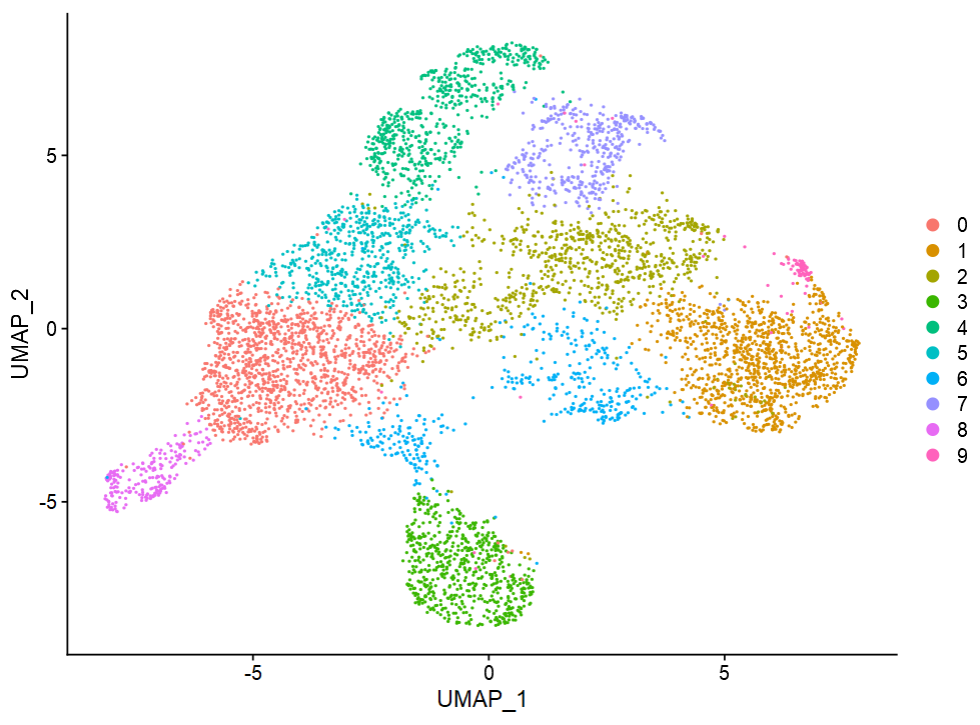
1.2 Seurat分析
- 使用Seurat包完成基本分析,主要包括UMAP降维与分群两步
|
|
2、下游分析
以下在jupyter notebook中执行
2.1 读入数据
- 参考之前scanpy学习笔记,导入Seurat对象为Anndata
|
|
|
|
|
|
|
|
- 读入velocyto分析的loom文件
|
|
- 合并上述两个数据
|
|

2.2 scvelo速率分析
- (1)统计每个cluster的spliced与unspliced比例
|
|
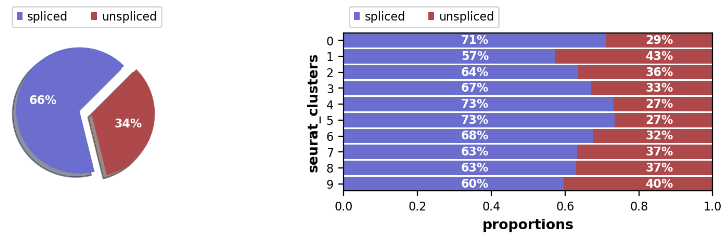
- (2)速率分析
|
|
- (3)速率分析结果可视化
|
|

|
|
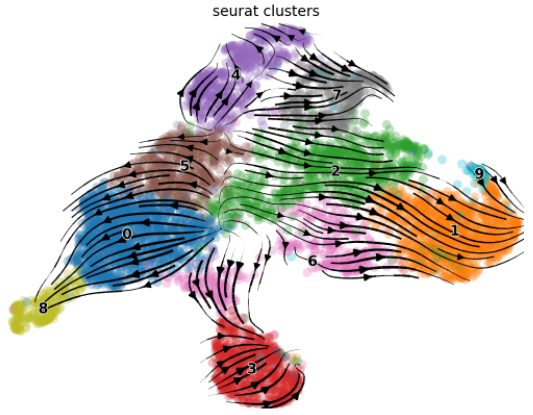
|
|
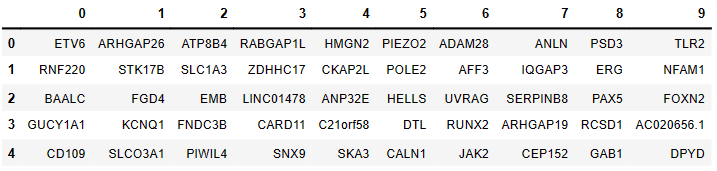
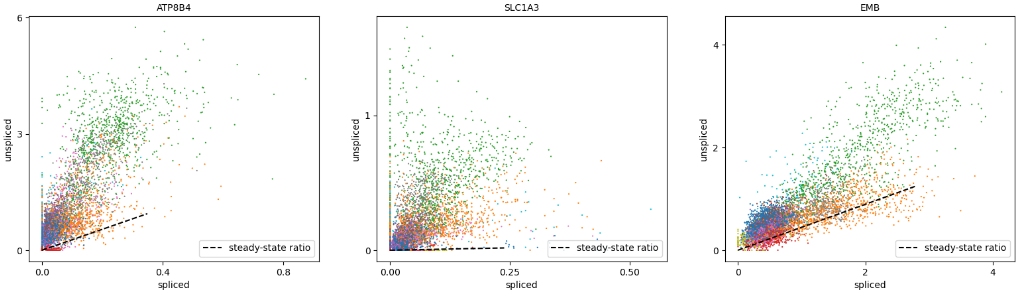
|
|

|
|

- (4)特定细胞群速率分析
|
|
|
|

|
|