SingleR包是在单细胞数据分析时用于细胞类型自动注释的常用工具。其基本原理是使用已有细胞标签的参考转录组数据集的表达谱,基于相似性原则注释未知单细胞数据的细胞类型。

1
2
3
4
|
library(Seurat)
library(SingleR)
library(celldex)
library(tidyverse)
|
1、示例数据#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
# download.file("https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz",
# "pbmc3k_filtered_gene_bc_matrices.tar.gz")
# untar("pbmc3k_filtered_gene_bc_matrices.tar.gz")
pbmc.data = Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
pbmc = CreateSeuratObject(counts = pbmc.data, project = "pbmc3k")
pbmc = NormalizeData(pbmc) %>%
FindVariableFeatures()
pbmc = ScaleData(pbmc) %>%
RunPCA() %>%
FindNeighbors(dims = 1:30) %>%
FindClusters(resolution = 0.1)
table(pbmc$seurat_clusters)
# 0 1 2 3 4
# 1201 684 450 351 14
|
2、基础用法#
- 参考(ref)/待注释(test)数据集可以是matrix矩阵格式或者SummarizedExperiment对象;
- ref数据集必须经log+normalization标准化处理;test数据集可以是raw count或者相关标准化。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
|
## test data
norm_count = GetAssayData(pbmc, slot="data")
dim(norm_count)
# [1] 32738 2700
## ref data
ref = HumanPrimaryCellAtlasData()
class(ref)
# [1] "SummarizedExperiment"
assays(ref)
# List of length 1
# names(1): logcounts
## 基础用法
pred = SingleR(test = norm_count,
ref = ref,
labels = ref$label.main,
de.method = "classic", #default
assay.type.test = "logcounts", #default
assay.type.ref = 1
)
head(pred)
# DataFrame with 5 rows and 4 columns
# scores labels delta.next pruned.labels
# <matrix> <character> <numeric> <character>
# 0 0.301403:0.700602:0.647725:... T_cells 0.2341943 T_cells
# 1 0.263423:0.660054:0.679101:... Monocyte 0.3058609 Monocyte
# 2 0.286134:0.653668:0.604933:... NK_cell 0.0535253 NK_cell
# 3 0.271754:0.749707:0.629452:... B_cell 0.2487106 B_cell
# 4 0.186824:0.387932:0.419774:... Platelets 0.1113118 Platelets
|
如上,SingleR默认为每一个细胞单独进行细胞注释。相关细节如下–
(1)labels标签提供ref数据集的细胞标签,一般celldex数据集均提供main与fine两种分辨率;
(2)assay.type.test/ref交代对应数据集的格式,默认均为’logcounts’。如果是SummarizedExperiment则设置数字交代logcounts所处assay的序号。
3、进阶用法#
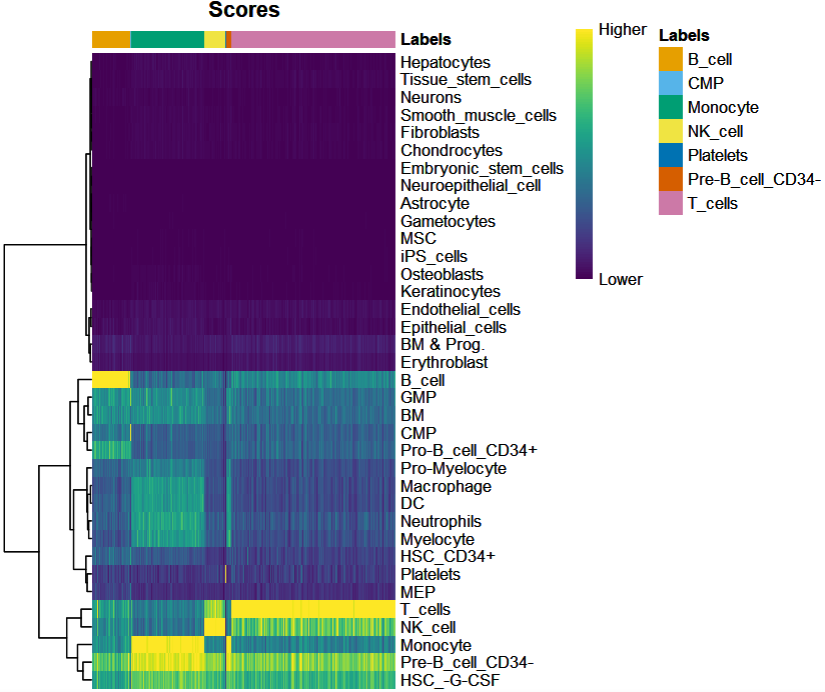
如下图的行标签表示参数数据集的全部细胞类型,列标签表示SingleR的注释结果。
1
2
3
4
5
6
|
pred = SingleR(test = norm_count,
ref = ref,
clusters = pbmc$seurat_clusters,
labels = ref$label.main)
pbmc$singleR_cluster = pred$labels[match(pbmc$seurat_clusters,
rownames(pred))]
|
- (3)若参考数据集为单细胞转录组,建议修改 de.method
1
2
3
4
|
pred = SingleR(test = norm_count,
ref = ref,
labels = ref$label.main,
de.method = "wilcox")
|
1
2
3
4
|
pred = SingleR(test = norm_count,
ref = ref,
labels = ref$label.main,
BPPARAM=MulticoreParam(8))
|
1
2
3
4
5
6
7
8
9
10
11
12
|
## 需确保训练数据与待测试数据的基因名一致
int_G = intersect(rownames(ref), rownames(norm_count))
# 训练模型
trained = trainSingleR(
ref = ref[int_G,],
labels = ref[int_G,]$label.main)
# 模型注释
pred = classifySingleR(
test = norm_count[int_G,],
trained = trained)
|
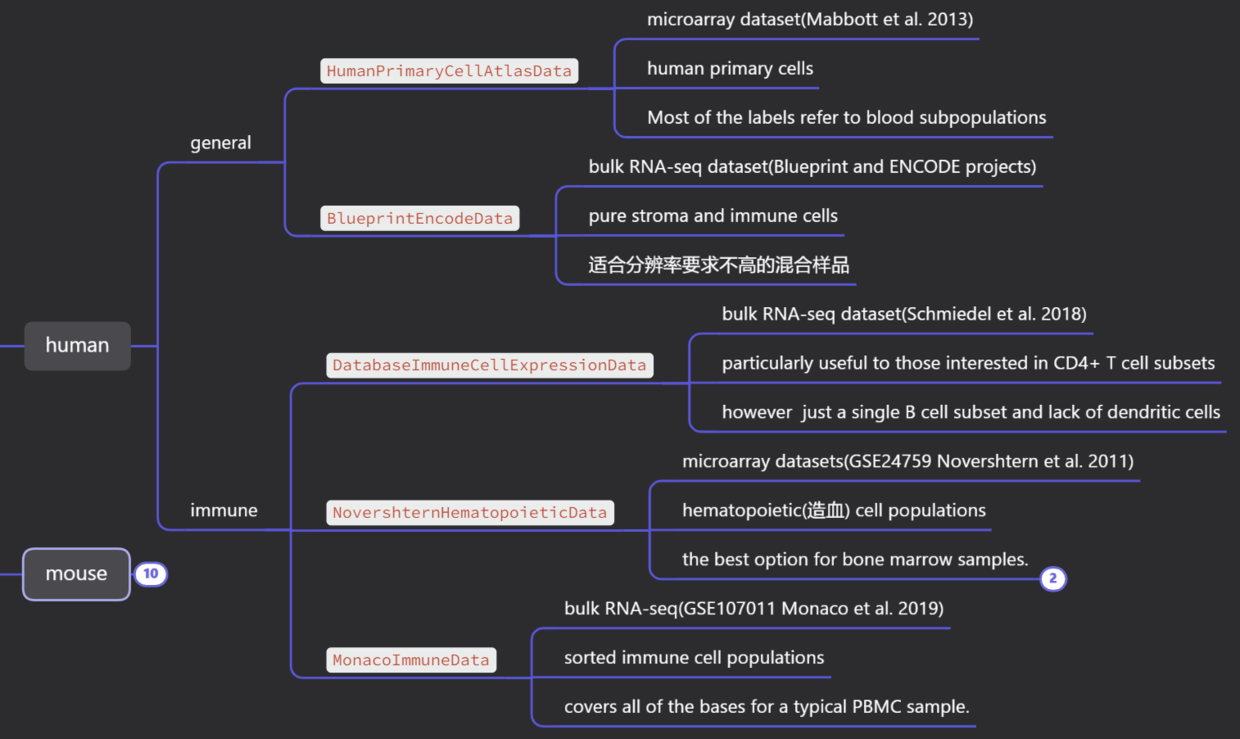
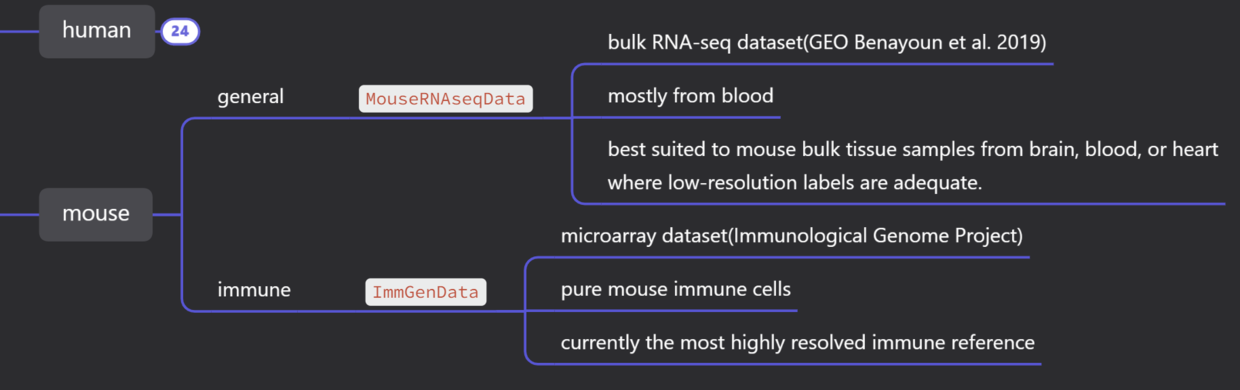
4、celldex概况#