inferCNV包可根据肿瘤组织相关的单细胞表达数据,推测肿瘤细胞的拷贝数变异情况,从而完成恶性细胞的鉴定。
1
2
3
|
BiocManager::install("infercnv")
library(infercnv)
# 暂时在window安装出现点问题,目前在linux使用conda安装、学习。
|
1、背景原理#
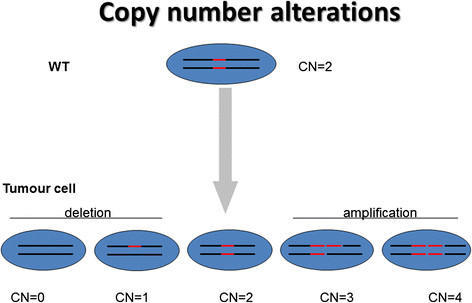
- 拷贝数变异是指指染色体上大于1 kb的DNA片段的扩增(amplification)或者减少(deletion),对基因的表达有很大的影响(扩增/降低)。而肿瘤恶性细胞通常伴随着拷贝数变异,通过影响相关基因的表达促进肿瘤发生。
- 在肿瘤单细胞数据分析过程中,肿瘤细胞类型的注释可通过tumor related marker gene的表达情况(是否高表达)做出判断。而inferCNV可以从拷贝数变异的角度进一步验证肿瘤细胞类型的注释。
- inferCNV的算法是在完成肿瘤微环境的细胞类型注释的基础之上,以“Normal”细胞的基因表达情况做对照,计算“tumor”-annotated 细胞中的某些染色体区域的基因表达是否发生明显的增多或减少,从而推测出细胞的拷贝数变异图谱(并可以进一步聚类),从而验证之前的注释结果。
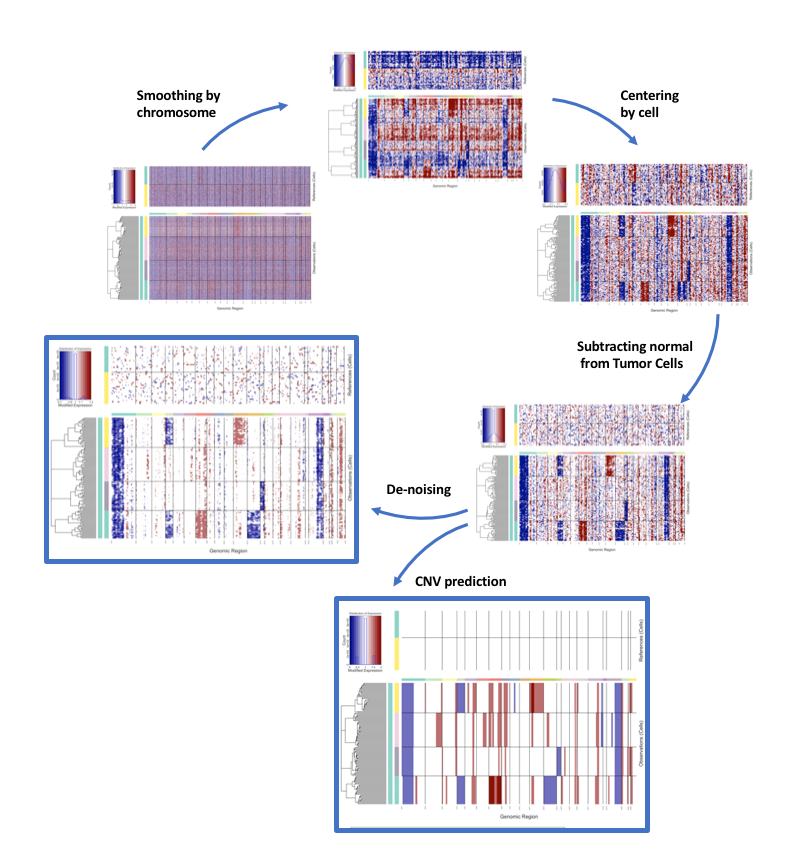
- inferCNV从计算步骤来说分为两大步:第一步根据Normal细胞对比,计算得到tumor-like细胞的CNV图谱(preliminary infercnv object);然后第二步是可选项,包括降噪处理和HMM预测,可分别得到两种结果。
2、输入数据#
infercnv分析主要需要三类数据,以R包自带的示例文件为例:
1
2
3
4
5
6
7
8
9
10
11
12
|
mat_dir = system.file("extdata", "oligodendroglioma_expression_downsampled.counts.matrix.gz", package = "infercnv")
raw_counts = data.table::fread(mat_dir, data.table = F)
rownames(raw_counts) = raw_counts[,1]
raw_counts = raw_counts[,-1]
raw_counts[1:4,1:4]
# MGH54_P16_F12 MGH54_P12_C10 MGH54_P11_C11 MGH54_P15_D06
# A2M 0 0.000 0.000 0.000
# A4GALT 0 0.000 0.000 0.000
# AAAS 0 37.008 30.935 21.011
# AACS 0 0.000 0.000 0.000
dim(raw_counts)
# [1] 10338 184
|
- (2)细胞类型注释文件
- 包含两列:第一列是细胞ID(对应表达矩阵的列名),第二列是细胞类型注释结果;
- 至少需要包含两种细胞类型:已知正常的细胞类型(免疫细胞、内皮细胞..)、可能为肿瘤细胞的细胞类型(肿瘤细胞、上皮细胞、成纤维细胞…);
- 此外由于肿瘤患者的异质性,不同病人来源的肿瘤细胞的拷贝数变异情况可能差别很大,因此可以在第二列的肿瘤细胞类型进行病源的注释,例如
tumor_P1,tumor_P2表示分别来自病人P1、P2的肿瘤细胞。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
anno_file_dir = system.file("extdata", "oligodendroglioma_annotations_downsampled.txt", package = "infercnv")
anno_file = data.table::fread(anno_file_dir, data.table = F, header = F)
head(anno_file)
dim(anno_file)
anno_file$V2 = stringr::str_split(anno_file$V2,"_",simplify = T)[,1]
head(anno_file)
# V1 V2
# 1 MGH54_P2_C12 Microglia/Macrophage
# 2 MGH36_P6_F03 Microglia/Macrophage
# 3 MGH53_P4_H08 Microglia/Macrophage
# 4 MGH53_P2_E09 Microglia/Macrophage
# 5 MGH36_P5_E12 Microglia/Macrophage
# 6 MGH54_P2_H07 Microglia/Macrophage
write.table(anno_file, quote = F, row.names = F, col.names = F,
sep = "\t", file = "anno_file.txt")
|
1
2
3
4
5
6
7
8
9
10
11
12
|
gene_order_file_dir = system.file("extdata", "gencode_downsampled.EXAMPLE_ONLY_DONT_REUSE.txt", package = "infercnv")
gene_order_file = data.table::fread(gene_order_file_dir, data.table = F, header = F)
head(gene_order_file)
# V1 V2 V3 V4
# 1 WASH7P chr1 14363 29806
# 2 LINC00115 chr1 761586 762902
# 3 NOC2L chr1 879584 894689
# 4 MIR200A chr1 1103243 1103332
# 5 SDF4 chr1 1152288 1167411
# 6 UBE2J2 chr1 1189289 1209265
write.table(gene_order_file, quote = F, row.names = F, col.names = F,
sep = "\t", file = "gene_order_file.txt")
|
3、示例分析#
3.1 构建对象#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
## 模板参数
infercnv_obj = CreateInfercnvObject(
#原始count矩阵
raw_counts_matrix=raw_counts_matrix,
#细胞类型注释信息
annotations_file=annotations_file,
#对应annotations_file里,认为是normal细胞的细胞类型
ref_group_names=c("celltype1","celltype2")
#基因坐标信息
gene_order_file=gene_order_file,
#指定上述两个文件的分隔符
delim="\t")
## 示例分析
infercnv_obj = CreateInfercnvObject(raw_counts_matrix=raw_counts,
annotations_file="anno_file.txt",
delim="\t",
gene_order_file="gene_order_file.txt",
ref_group_names=c("Microglia/Macrophage",
"Oligodendrocytes (non-malignant)"))
|
3.2 变异分析#
默认计算得到preliminary infercnv object,可分别设置参数交代是否进行进一步降噪(de-noising)或者CNV的HMM预测。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
|
## 模板参数
infercnv_obj = infercnv::run(
#上一步构建的infercnv对象
infercnv_obj,
#筛选基因的阈值:基因在所有细胞的平均表达量
# use 1 for smart-seq, 0.1 for 10x-genomics
cutoff=1,
#存储输出结果的文件夹名(每一步的中间文件都会保存)
out_dir="output_dir",
#是否将肿瘤细胞按照病源(病人之间异质性)分群计算CNV图谱
cluster_by_groups=T,
#是否降噪处理
denoise=T,
#是否利用HMM算法预测CNV状态
HMM=T,
#使用的线程数
num_threads = 8)
## 示例分析
infercnv_obj = infercnv::run(infercnv_obj,
cutoff=1,
out_dir="infer_out",
cluster_by_groups=TRUE,
denoise=T,
HMM=F)
|
3.3 结果解读#
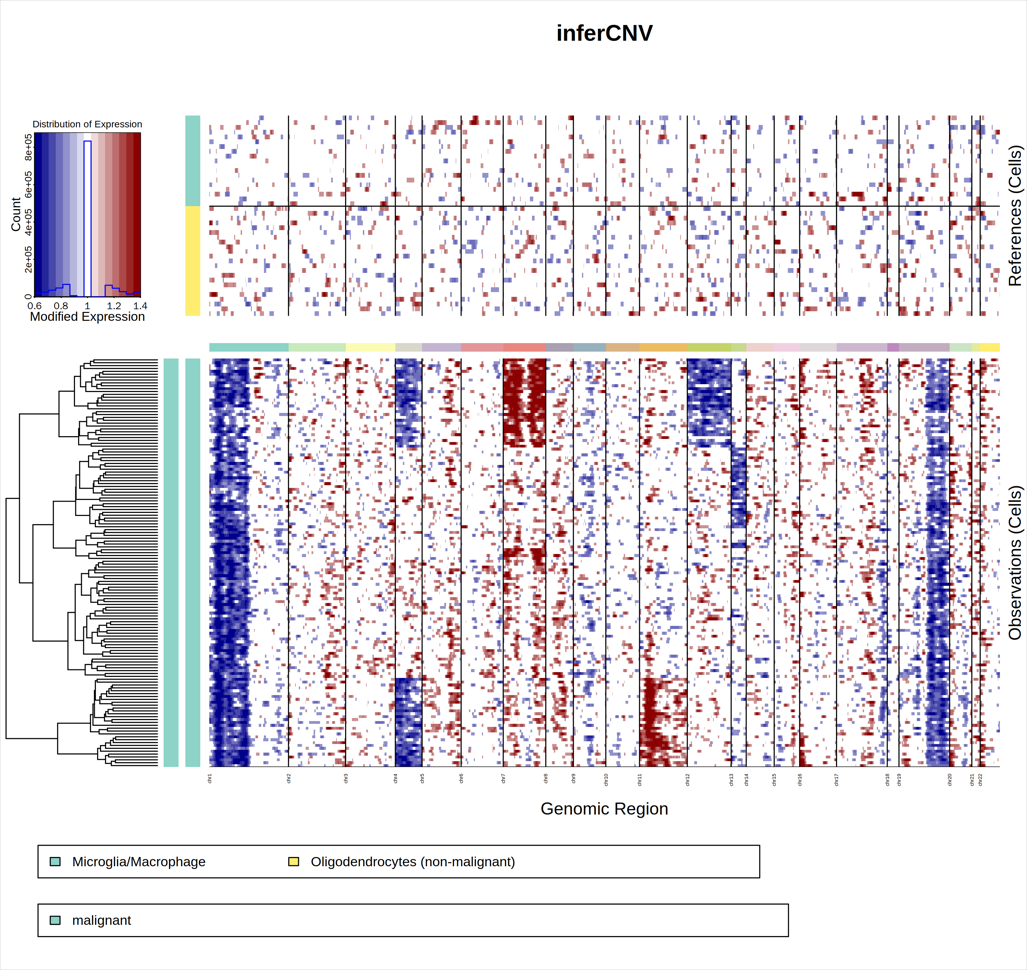
如下图为infercnv的分析结果可视化呈现,可分为3部分:上半部分热图、下半部分热图以及左上角的图例
- 首先关于左上角的图例:(0,0.5,1,1.5,2)分别表示相对于Normal细胞的染色体区域基因表达量的倍数,红色表示该区域基因量相对增多,蓝色表示该区域基因量相对减少。柱子的长度表示对应区域的多少;
- 上半部分的热图:表示指定为Normal细胞的表达分布情况,正常情况下应该都是白色,没有明显集中的CNV区域;
- 下半部分的热图:相对于上半部分的Normal cell,计算的得到的每个tumor-like细胞的CNV图谱;然后根据所有细胞的相似性进行树状图聚类。