WGCNA是适用于大批量样本的array或者Bulk RNAseq数据的加权基因共表达网络分析。由于单细胞数据的稀疏性,不适用于WGCNA直接分析。hdWGCNA包基于WGCNA包提供了一种针对scRNAseq数据的加权基因共表达网络分析策略。
官方教程:https://smorabit.github.io/hdWGCNA/articles/basic_tutorial.html
1、加载包并准备示例数据#
1
2
3
4
5
6
7
8
9
10
11
12
13
|
#devtools::install_github('smorabit/hdWGCNA', ref='dev')
library(hdWGCNA)
library(WGCNA)
library(Seurat)
library(clustree)
library(dplyr)
library(patchwork)
# 示例数据
# https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
list.files("filtered_gene_bc_matrices/hg19/")
# [1] "barcodes.tsv" "genes.tsv" "matrix.mtx"
|
2、创建Seurat对象并预处理#
执行Seurat的常规流程,包括:创建seurat对象—标准化、归一化—高变基因—降维、聚类分群—鉴定细胞类型(optional)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
sce = Read10X(data.dir = "filtered_gene_bc_matrices/hg19/") %>% CreateSeuratObject()
# 32738 features across 2700 samples
sce = sce %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData()
sce = sce %>%
RunPCA() %>%
RunUMAP(dims = 1:30) %>% #RunTSNE
FindNeighbors(dims = 1:30) %>%
FindClusters(resolution = c(0.01, 0.05, 0.1, 0.2, 0.3, 0.5,0.8,1))
clustree(sce@meta.data, prefix = "RNA_snn_res.")
#colnames(sce@meta.data)
table(sce$RNA_snn_res.0.1)
# 0 1 2 3
# 1195 698 448 359
Idents(sce) = "RNA_snn_res.0.1"
##如上按照分辨率为0.1的分群结果进行后续的分析
|
3、创建misc slot,挑选基因#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
seurat_obj = SetupForWGCNA(
sce,
gene_select = "fraction", # the gene selection approach
fraction = 0.05, # fraction of cells that a gene needs to be expressed in order to be included
wgcna_name = "PBMC" # the name of the hdWGCNA experiment
)
# gene_select参数设置过滤基因的方法,还有是
## (1) "variable" : use the genes stored in the Seurat object’s VariableFeatures.
## (2) "custom" : use genes that are specified in a custom list.
str(seurat_obj@misc)
# List of 2
# $ active_wgcna: chr "PBMC"
# $ PBMC :List of 2
# ..$ wgcna_group: chr "all"
# ..$ wgcna_genes: chr [1:3855] "NOC2L" "HES4" "ISG15" "TNFRSF4" ...
|
将多个细胞表达情况(KNN)合并为一个metacell,可避免单细胞数据的稀疏性。
“多个细胞"需要是来自同一种细胞类型/分群,可通过group.by参数定义依据。
k参数:in general a lower number for k can be used for small datasets. We generally use k values between 20 and 75.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
|
seurat_obj <- MetacellsByGroups(
seurat_obj = seurat_obj,
group.by = c("RNA_snn_res.0.1"), # specify the columns in seurat_obj@meta.data to group by
k = 25, # nearest-neighbors parameter
max_shared = 10, # maximum number of shared cells between two metacells
ident.group = 'RNA_snn_res.0.1' # set the Idents of the metacell seurat object
)
seurat_obj <- NormalizeMetacells(seurat_obj)
seurat_obj@misc$PBMC$wgcna_metacell_obj
# 32738 features across 1524 samples within 1 assay
seurat_obj@misc$PBMC$wgcna_metacell_obj@assays$RNA@counts[1:4,1:8]
# 4 x 8 sparse Matrix of class "dgCMatrix"
# 0_1 0_2 0_3 0_4 0_5 0_6 0_7 0_8
# MIR1302-10 . . . . . . . .
# FAM138A . . . . . . . .
# OR4F5 . . . . . . . .
# RP11-34P13.7 . . . . . . . .
seurat_obj@misc$PBMC$wgcna_metacell_obj@meta.data %>% head
# orig.ident nCount_RNA nFeature_RNA RNA_snn_res.0.1
# 0_1 0 2618.96 5880 0
# 0_2 0 2486.00 5858 0
# 0_3 0 1340.56 4306 0
# 0_4 0 2112.12 5388 0
# 0_5 0 2839.04 6073 0
# 0_6 0 2198.44 5407 0
|
5、共表达网络分析#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
##(1) 选择一种/多种细胞类型,进行WGCNA分析
seurat_obj <- SetDatExpr(
seurat_obj,
group_name = c(0,1,2,3),
group.by='RNA_snn_res.0.1' # the metadata column containing the cell type info. This same column should have also been used in MetacellsByGroups
)
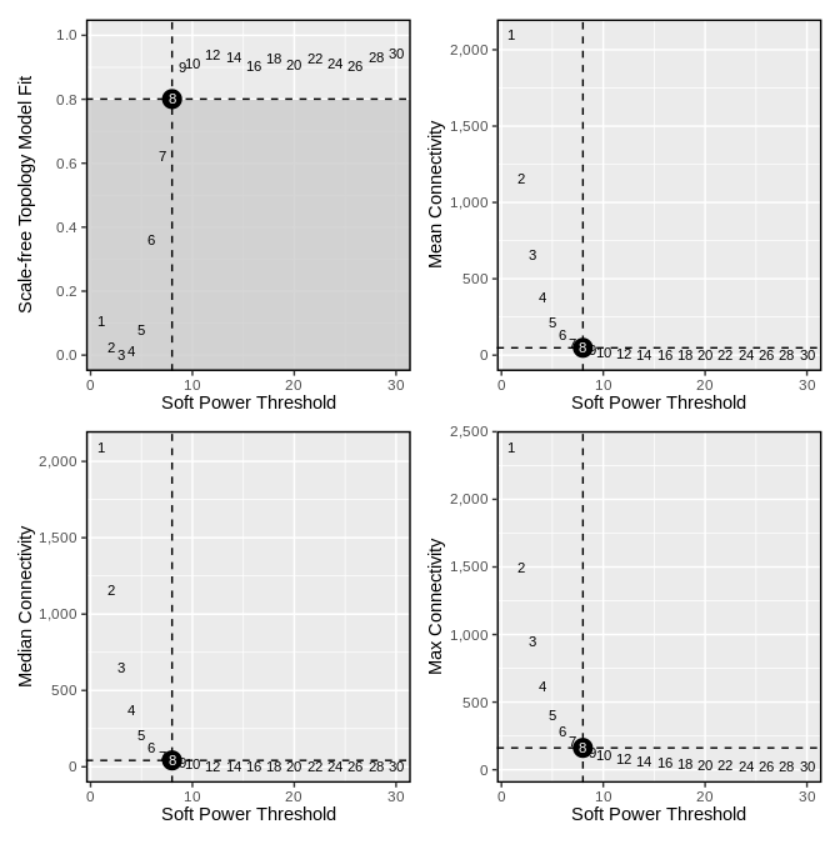
##(2) 选择合适的软阈值
seurat_obj <- TestSoftPowers(
seurat_obj,
setDatExpr = FALSE, # set this to FALSE since we did this above
)
plot_list <- PlotSoftPowers(seurat_obj)
wrap_plots(plot_list, ncol=2)
# power_table <- GetPowerTable(seurat_obj)
# head(power_table)
|
1
2
3
4
5
6
|
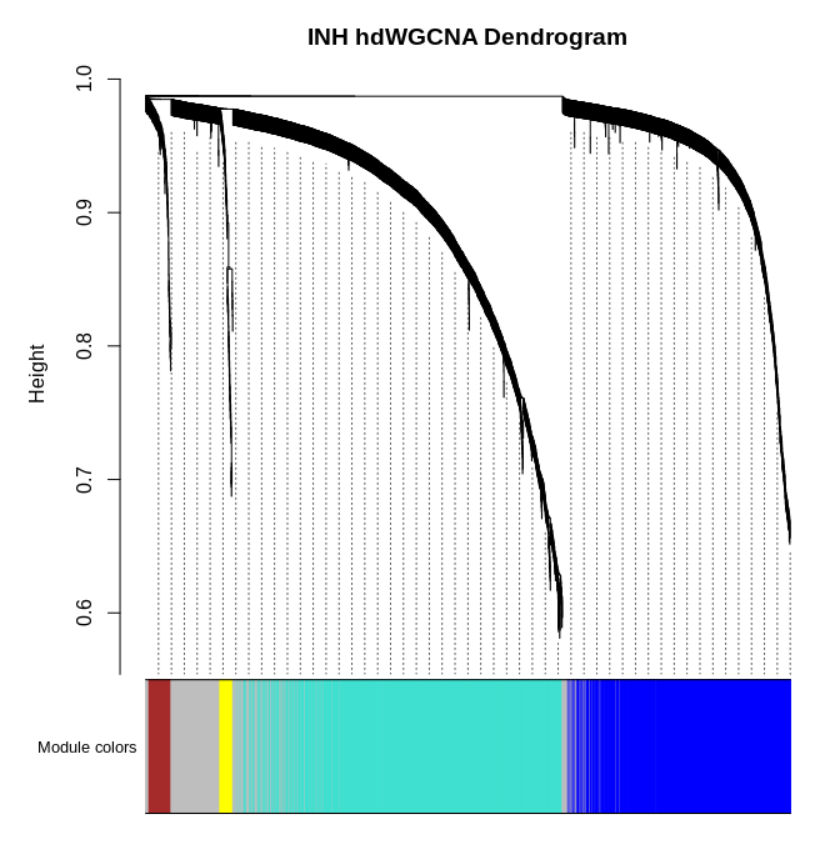
##(3) 根据上图选择合适的软阈值构建网络
seurat_obj <- ConstructNetwork(
seurat_obj, soft_power=8,
setDatExpr=FALSE
)
PlotDendrogram(seurat_obj, main='INH hdWGCNA Dendrogram')
|
6、模块特征值与hub基因分析#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
seurat_obj@misc$PBMC$wgcna_modules %>% head
# gene_name module color
# NOC2L NOC2L grey grey
# HES4 HES4 turquoise turquoise
# ISG15 ISG15 turquoise turquoise
# TNFRSF4 TNFRSF4 blue blue
# SDF4 SDF4 blue blue
# UBE2J2 UBE2J2 blue blue
table(seurat_obj@misc$PBMC$wgcna_modules$module)
# grey turquoise blue brown yellow
# 588 1786 1272 133 76
# modules <- GetModules(seurat_obj)
# head(modules)
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
seurat_obj <- ModuleEigengenes(
seurat_obj
)
seurat_obj@misc$PBMC$MEs %>% head
# blue grey brown turquoise yellow
# AAACATACAACCAC-1 4.755930 0.2424180 2.6565029 -5.832575 -2.0906948
# AAACATTGAGCTAC-1 2.388492 1.3106515 -0.3341197 1.172415 8.8764667
# AAACATTGATCAGC-1 5.807490 2.9008702 0.8873175 -1.132285 -1.8973713
# AAACCGTGCTTCCG-1 -6.662387 -0.3072945 -1.5451723 14.692779 0.1159869
# AAACCGTGTATGCG-1 -16.098322 -1.6676024 8.7666564 -4.131657 -0.9371790
# AAACGCACTGGTAC-1 2.064300 -0.1415680 -1.2313300 -2.824847 -1.1698044
# MEs <- GetMEs(seurat_obj, harmonized=FALSE)
# head(MEs)
|
- (3)鉴定模块内hub基因– 基因表达与模块特征值的具有高相关性
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
|
seurat_obj <- ModuleConnectivity(
seurat_obj
)
seurat_obj@misc$PBMC$wgcna_modules[,c(-1,-2,-3)] %>% head
# kME_blue kME_grey kME_brown kME_turquoise kME_yellow
# NOC2L 0.02851618 0.08360662 0.02041163 0.003048238 0.038497122
# HES4 -0.24225802 0.06869209 -0.08284058 0.117744840 0.326692215
# ISG15 -0.29705821 0.08027215 -0.07240235 0.351103776 0.314836382
# TNFRSF4 0.16100218 0.08589770 0.13010946 -0.002353201 -0.090182722
# SDF4 0.01120696 0.13914234 0.07918151 0.040070556 0.003869467
# UBE2J2 0.03178884 0.07651548 0.04663831 0.008615164 -0.006942615
# p <- PlotKMEs(seurat_obj, ncol=2)
# p
hub_df <- GetHubGenes(seurat_obj, n_hubs = 10)
hub_df
# gene_name module kME
# 1 RPS27 blue 0.7207118
# 2 RPS12 blue 0.7258250
# 3 CCL5 brown 0.4934812
# 4 B2M brown 0.5294537
# 5 LGALS1 turquoise 0.6477877
# 6 TYROBP turquoise 0.6542149
# 7 HLA-DRA yellow 0.5817169
# 8 HLA-DPA1 yellow 0.5894472
## 计算每个细胞对于每个模块hub基因的表达活性(module score),可使用seurat包或者Ucell包
seurat_obj <- ModuleExprScore(
seurat_obj,
n_genes = 25,
method='Seurat'
)
seurat_obj@misc$PBMC$module_scores %>% head
## 计算每个细胞对于每个模块hub基因的表达活性(module score),可使用seurat包或者Ucell包
# blue brown turquoise yellow
# AAACATACAACCAC-1 3.380281 0.74403040 0.3806462 -0.18699177
# AAACATTGAGCTAC-1 3.035111 0.31949694 0.4019337 1.06061130
# AAACATTGATCAGC-1 2.769994 0.28652286 0.5089602 -0.30358276
# AAACCGTGCTTCCG-1 2.080403 0.07190668 2.6016017 0.45921797
# AAACCGTGTATGCG-1 1.434566 1.07243443 0.9849420 0.14309843
# AAACGCACTGGTAC-1 2.999822 0.25594526 0.6041499 -0.02860217
# library(UCell)
# seurat_obj <- ModuleExprScore(
# seurat_obj,
# n_genes = 25,
# method='UCell'
# )
# seurat_obj@misc$PBMC$module_scores %>% head
|
7、结果可视化#
1
2
3
4
5
6
|
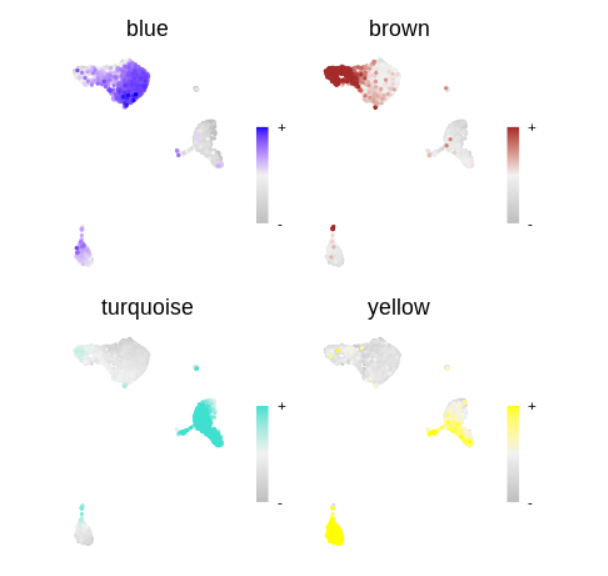
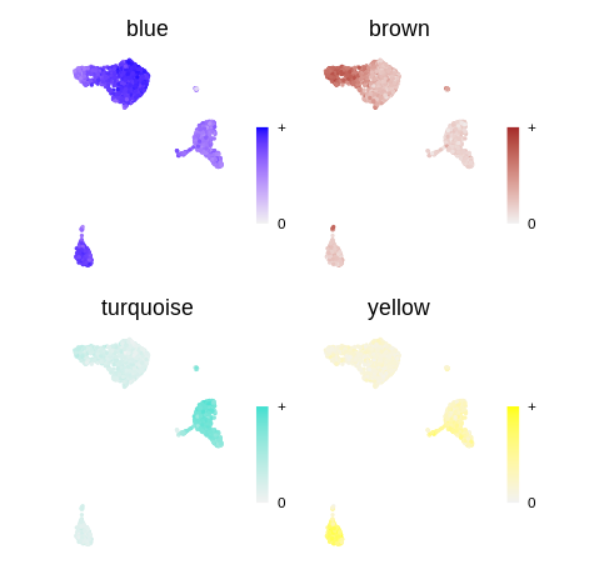
plot_list <- ModuleFeaturePlot(
seurat_obj,
features='MEs', # plot the hMEs
order=TRUE # order so the points with highest hMEs are on top
)
wrap_plots(plot_list, ncol=2)
|
1
2
3
4
5
6
7
8
|
seurat_obj@misc$PBMC$module_scores %>% head
plot_list <- ModuleFeaturePlot(
seurat_obj,
features='scores', # plot the hub gene scores
order='shuffle', # order so cells are shuffled
ucell = TRUE # depending on Seurat vs UCell for gene scoring
)
wrap_plots(plot_list, ncol=2)
|
- 将细胞对于模块的特征值,整合到seurat的meta.data中
1
2
3
4
5
6
7
8
9
10
|
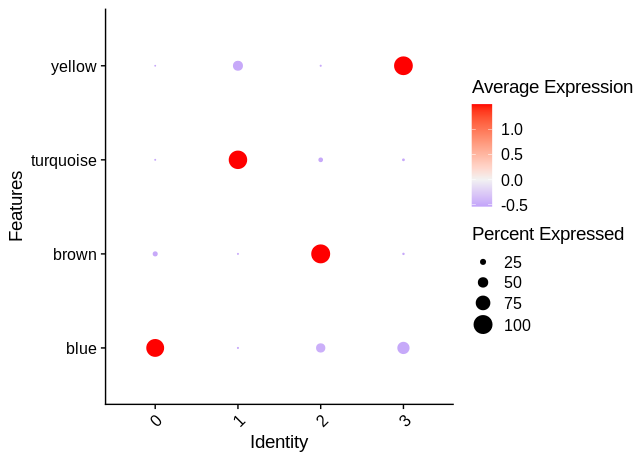
MEs <- GetMEs(seurat_obj)
mods <- colnames(MEs); mods <- mods[mods != 'grey']
seurat_obj@meta.data <- cbind(seurat_obj@meta.data, MEs)
p <- DotPlot(seurat_obj, features = mods, group.by = 'RNA_snn_res.0.1')
p <- p +
coord_flip() +
RotatedAxis() +
scale_color_gradient2(high='red', mid='grey95', low='blue')
p
|