Paper:https://www.nature.com/articles/s41587-019-0068-4
github:https://github.com/dpeerlab/Palantir
tutorial:https://nbviewer.org/github/dpeerlab/Palantir/blob/master/notebooks/Palantir_sample_notebook.ipynb
简介:Palantir是一个2019年在nature biotechnology提出的用于单细胞数据轨迹推断的Python工具包。根据官方教程,简单学习用法如下。
0、安装包与准备数据#
1)安装python包#
- 根据前期探索,不建议直接使用pip安装(存在bug),而是下载github的最新源码后安装。
1
2
3
|
unzip Palantir-master
cd Palantir-master
pip install .
|
2)示例数据#
- 需要准备**.h5ad**的单细胞数据(count表达矩阵)格式,官方提供示例数据可直接下载。
1
|
wget https://dp-lab-data-public.s3.amazonaws.com/palantir/marrow_sample_scseq_counts.h5ad
|
Seurat对象转为h5ad格式,参考笔记:***
1、加载包与环境#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
import palantir
import scanpy as sc
import numpy as np
import pandas as pd
import os
# Plotting
import matplotlib
import matplotlib.pyplot as plt
import seaborn as sns
import warnings
# Inline plotting
%matplotlib inline
sns.set_style('ticks')
matplotlib.rcParams['figure.figsize'] = [4, 4]
matplotlib.rcParams['figure.dpi'] = 100
matplotlib.rcParams['image.cmap'] = 'Spectral_r'
warnings.filterwarnings(action="ignore", module="matplotlib", message="findfont")
# Reset random seed
np.random.seed(5)
|
2、导入数据与预处理#
1
2
3
4
5
6
7
8
9
10
11
12
|
## (1) 导入数据
ad = sc.read('marrow_sample_scseq_counts.h5ad')
ad
# AnnData object with n_obs × n_vars = 4142 × 16106
# 4142个细胞,16106个基因
## (2) 标准化
sc.pp.normalize_per_cell(ad)
palantir.preprocess.log_transform(ad)
## (3) 高变基因
sc.pp.highly_variable_genes(ad, n_top_genes=1500, flavor='cell_ranger')
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
|
## (1) scanpy
#PCA降维
sc.pp.pca(ad)
#UMAP降维
sc.pp.neighbors(ad)
sc.tl.umap(ad)
## (2) Seurat
seurat_pca = pd.read_csv("marrow_sample_scseq_pca.csv", index_col=0).values
ad.obsm['X_pca'] = seurat_pca
seurat_umap = pd.read_csv("marrow_sample_scseq_umap.csv", index_col=0).values
ad.obsm['X_umap'] = seurat_umap
ad
# AnnData object with n_obs × n_vars = 4142 × 16106
# obs: 'n_counts'
# var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
# uns: 'hvg', 'pca', 'neighbors', 'umap'
# obsm: 'X_pca', 'X_umap'
# varm: 'PCs'
# obsp: 'distances', 'connectivities'
pca_projections = pd.DataFrame(ad.obsm['X_pca'], index=ad.obs_names)
umap = pd.DataFrame(ad.obsm['X_umap'], index=ad.obs_names)
# umap可视化
sc.pl.embedding(ad, basis='umap')
|
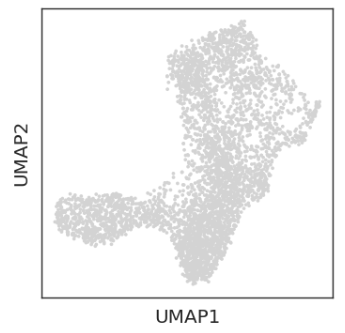
3、轨迹推断分析#
1
2
3
4
5
6
7
8
9
|
## (1)Run diffusion maps
dm_res = palantir.utils.run_diffusion_maps(pca_projections, n_components=5)
ms_data = palantir.utils.determine_multiscale_space(dm_res)
ad.layers['MAGIC_imputed_data'] = palantir.utils.run_magic_imputation(ad, dm_res)
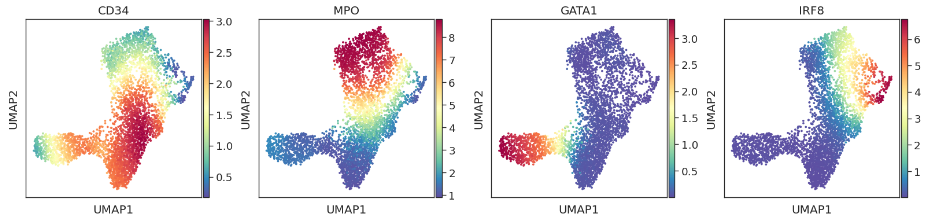
#基因表达量可视化
sc.pl.embedding(ad, basis='umap', layer='MAGIC_imputed_data',
color=['CD34', 'MPO', 'GATA1', 'IRF8'])
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
## (2)轨迹推断
# 定义发育起点(required)
start_cell = 'Run5_164698952452459'
# 定义可能的发育终点(optional)
terminal_states = pd.Series(['DC', 'Mono', 'Ery'],
index=['Run5_131097901611291',
'Run5_134936662236454',
'Run4_200562869397916'])
# 轨迹推断
pr_res = palantir.core.run_palantir(ms_data, start_cell, num_waypoints=500, terminal_states=terminal_states.index)
pr_res.branch_probs.columns = terminal_states[pr_res.branch_probs.columns]
# 基本可视化
palantir.plot.plot_palantir_results(pr_res, umap)
|
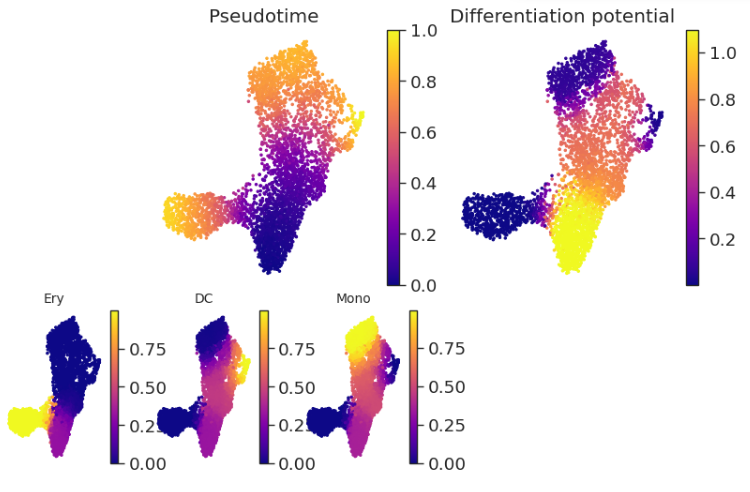
4、结果可视化#
1
2
|
cells = ['Run5_164698952452459', 'Run5_170327461775790', 'Run4_121896095574750', ]
palantir.plot.plot_terminal_state_probs(pr_res, cells)
|
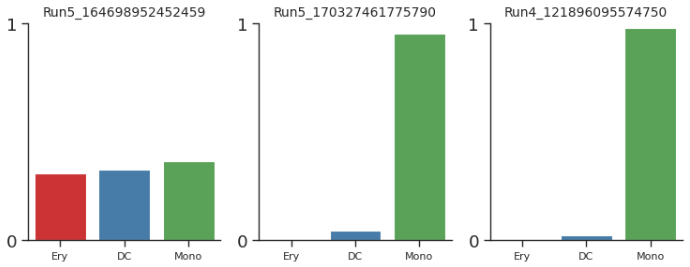
1
|
palantir.plot.highlight_cells_on_tsne(umap, cells)
|
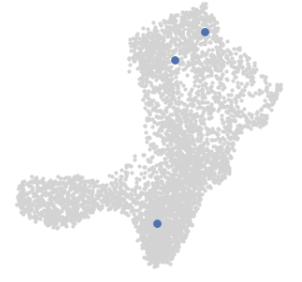
1
2
3
4
5
6
7
|
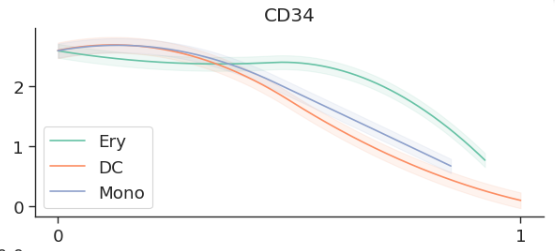
genes = ['CD34', 'MPO', 'GATA1', 'IRF8']
imp_df = pd.DataFrame(ad[:, genes].layers['MAGIC_imputed_data'],
index=ad.obs_names, columns=genes)
gene_trends = palantir.presults.compute_gene_trends( pr_res, imp_df.loc[:, genes])
## 线图可视化
palantir.plot.plot_gene_trends(gene_trends)
|
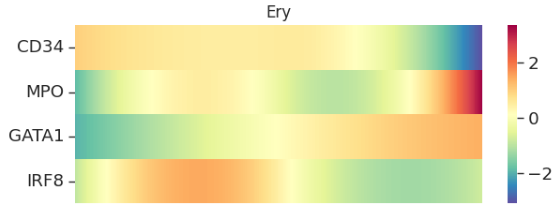
1
2
|
## 热图可视化
palantir.plot.plot_gene_trend_heatmaps(gene_trends)
|
1
2
3
4
5
6
7
8
9
|
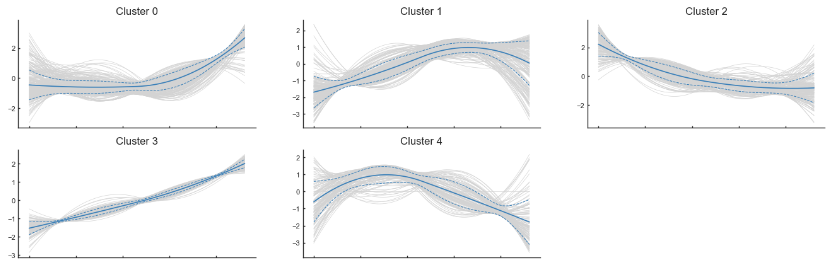
genes = ad.var_names[:1000]
imp_df = pd.DataFrame(ad[:, genes].layers['MAGIC_imputed_data'],
index=ad.obs_names, columns=genes)
gene_trends = palantir.presults.compute_gene_trends(pr_res,
imp_df.iloc[:, 0:1000], ['Ery'])
trends = gene_trends['Ery']['trends']
gene_clusters = palantir.presults.cluster_gene_trends(trends)
palantir.plot.plot_gene_trend_clusters(trends, gene_clusters)
|
最后如原文所述, 由于自身算法限制,本工具可能不太适合应用于肿瘤细胞生长、迁移等存在dedifferetiate或者trans-differentiate to earlier transcriptional state的发育轨迹推断
