SCENIC全称Single-Cell rEgulatory Network Inference and Clustering,即转录因子分析,用于构建GRN,gene regulatory network基因调控网络。2017年于Nat Methods首次发表R包版本(https://doi:10.1038/nmeth.4463.),后又于2020年NATURE PROTOCOLS发表python版本(https://doi.org/10.1038/s41596-020-0336-2),分析速度得到大幅度提升,而分析方法基本相同。
这篇笔记首先简单了解下分析步骤与原理,然后参考文章提供代码进行实操
1、TF转录因子
-
转录因子(Transcription factor)是指能够识别、结合在某基因上游特异核苷酸序列上的蛋白质;
-
这类蛋白通过介导RNA聚合酶与DNA模板的结合,从而调控下游靶基因的转录;也可以和其它转录因子形成转录因子复合体来影响基因的转录。

-
转录因子特异识别、结合的DNA序列称为转录因子结合位点(Transcription factor binding site,TFBS),长度在5~20bp范围。由于同一个TF可以调控多个,而其具体结合到每个靶基因的TFBS不完全相同,但具有一定的保守性。
-
基于此,使用motif来概括TFBS的特征序列,具体是使用PWM(Postion Weight Matrix)表示。如下例图,即有的位点是某一种特定碱基,而有的位点是多种可能。

- 同一个基因上游可能存在多种motif,受到多个TF调控;而同一个TF也可能识别多种motif,从而形成复杂的基因表达转录调控网络。
2、SCENIC分析步骤
2.1 单细胞数据预处理
- 输入:单细胞测序数据;输出:单细胞表达矩阵
这一步主要就是读入单细胞数据,过滤低质量的细胞、低表达的基因;然后将单细胞count表达矩阵(行名是细胞,列名是基因)存储为csv格式(或者loom);
文章建议使用Scanpy预处理,因为更适合大的单细胞数据集。但个人还是更习惯于Seurat的流程。
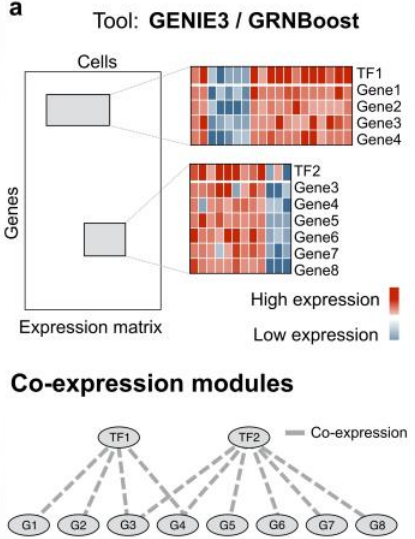
2.2 网络推断Network inference
- 输入:单细胞表达矩阵,转录因子列表;输出:TF-gene.csv文件。第1列是转录因子,第2列是共表达的基因,第3列是共表达强度(weight)
针对转录因子列表里的每一个转录因子,探索在单细胞表达矩阵中哪些基因与之存在共表达关系。
通过基于树(tree-based)的回归模型,根据转录因子的表达去预测基因的表达情况。具体算法可选择GRNBoost2或者GENIE3,前者的计算效率会更高。
2.3 得到TF-regulon
- 输入:上一步得到的TF-gene.csv,motif-gene注释数据,motif-TF注释数据,单细胞表达数据
- 输出:motif富集结果或者regulon数据
分为如下几个子步骤
(1) Module generation
首先根据第二步得到的TF-gene.csv文件,计算中TF -module(one Translate factor and its target genes)。scenic提供下面三种思路计算得到。
- Based on a percentile score; the default is to select sets based on the 75th or 90th percentile of a factor’s targets.
- Based on the top N targets for each factor, with a default of 50 targets.
- Based on the top N regulators for a target; the default settings select sets of 5, 10, and 50 regulators.
- 按照如上默认值设置的话,有 2 + 1 + 3 = 6种思路的module结果。所以在最终得到的结果里,可能同一个TF有多个module。
根据调控基因与转录因子的相关性方向可分为 activating module(ρ ≥ +0.03)与 repressing module(ρ ≤ −0.03)。由于负相关模块(TF介导对靶基因的转录抑制)的数量一般较少,默认后续分析只保留正相关的activating module进行后续分析。
最后可限制module所包含的最少基因数,默认为20。
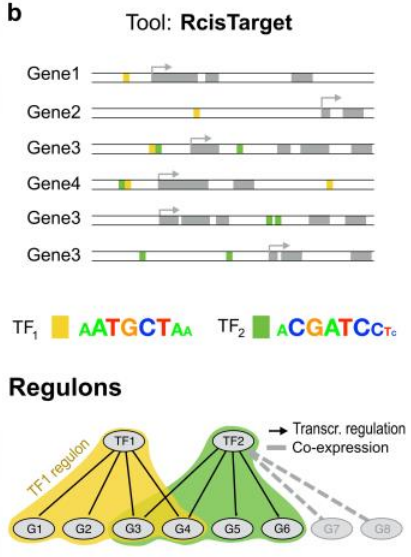
(2) Motif enrichment
在提供的motif-gene注释文件中,提供了每个motif的基因排名,即哪些基因的上游区域含有最类似该motif的区域。
基于此计算出每个module中所富集的motif有哪些。然后根据motif-TF注释数据,判断module 所富集到的motif是否为该module TF的motif。再然后,通过module的motif预测模块中真正的靶基因,去除其中的假阳性或者间接靶点。
最后通过合并同一个TF的所有保留基因,作为一个Regulon(one TF and its target gene)
2.4 Regulon活性评分
基于上一步得到的所有Regulon(可认为是Gene Set),结合单细胞表达矩阵去评价每一个细胞对于每一个Regulon的表达程度评分。
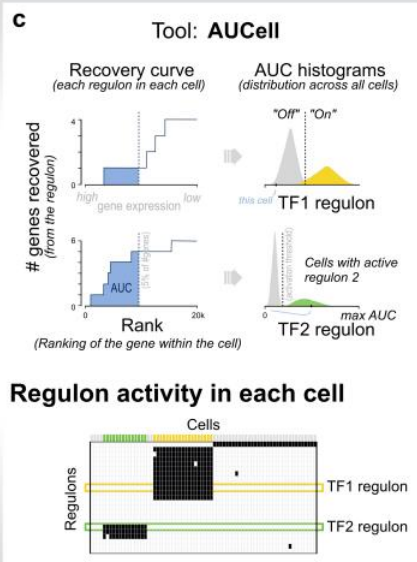
不同细胞对于同一Regulon的表达活性是可以比较的,但同一细胞的不同Regulon的表达与否是不好比较的。
文章提供一种分析思路,将Regulon的活性得分二值化,即是否激活(On/Off)。这主要通过对那些具有 bimodal distribution双峰分布曲线的Regulon进行混合高斯分布拟合,确定合适的分割阈值,从而将细胞划分为两种状态。
原先参考代码是pySCENIC文章里提供的示范代码;如今版本以及数据库均已更新,现主要参考官方手册:
3、pySCENIC代码实操
原文的预处理步骤是使用Scanpy进行单细胞预处理的,我还是比较熟悉Seurat流程,因此对文章给的代码有所删改。
3.1 准备conda环境
|
|
3.2 下载参考数据
-
(1)motif–gene注释数据(genome ranking database)
https://resources.aertslab.org/cistarget/databases/ 提供了不同物种以及不同基因组版本的注释数据,可根据需要选择;
|
|
-
(2)motif–TF注释数据
https://resources.aertslab.org/cistarget/motif2tf/ 提供了不同物种的注释数据,可根据需要选择;
|
|
-
(3)转录因子列表
https://resources.aertslab.org/cistarget/tf_lists/ 提供了不同物种的注释数据,可根据需要选择;
|
|
3.3 示例单细胞表达矩阵
|
|
Note:
- 上面输出、保存的表达矩阵行名是基因,列名是细胞;
- 而SCENIC分析需要的表达矩阵格式为:行名是细胞,列名是基因;
- 因此可在之后pyscenic分析时添加参数 –transpose,进行转置。
3.4 scenic分析(命令行)
分别对应上述的2.2~2.4简介
|
|
(1) pyscenic grn
|
|
(2) pyscenic ctx
|
|
(3) pyscenic aucell
|
|
- 将AUCell结果二值化:on/off。需要进入python环境
|
|
Note :
(1)输入单细胞表达矩阵也支持loom格式;输出结果中也支持不同类型的数据格式,具体可参看命令帮助文档 。
(2)官方手册中也提供了多种不同使用方法,例如在python中的分析流程;docker环境中的使用方法。