单细胞转录组测序技术历经10余年的发展,目前以高通量的Droplet技术广为应用。其中以10X Genomics公司开发的实验及分析系统最为流行。如下简单学习测序原理、结果以及上游比对流程。
1、测序原理#
(1)如下图所示,首先每个凝胶微珠(Gel Beads)表面提前接上大量定制的DNA片段。该片段主要由3部分组成:10x Barcode、UMI以及Poly(dT)尾巴。
- 10x Barcode:长度为16的核苷酸序列,用于区分不同的微珠(细胞);
- UMI:长度为10的核苷酸序列,用于区分不同的cDNA分子;
- Poly(dT):用于结合mRNA的Poly(A)尾巴;
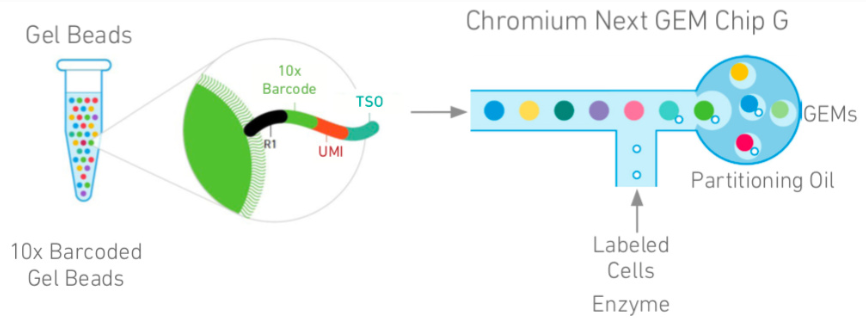
(2)理想情况下,一个微珠会捕捉到一个细胞,并形成油包水的封闭体系。然后在酶反应作用下,裂解细胞,释放的mRNA分子结合到微珠表面的DNA片段上;最后逆转录形成相应的cDNA分子。

(3)提取所有水相里的cDNA,进行PCR扩增,之后接入illumina高通量双端测序分析。
- Read 1,即R1,通常表示长度为26的10X barcode与UMI标签序列;
- Read 2,即R2,通常为长度为98的mRNA测序片段。
- 参考https://www.biostars.org/p/9529864/#9529942,如果下载的R1、R2文件为150bp长度,也可直接分析。
UMI标签主要用于消除PCR扩增带来的误差,即表示细胞裂解是结合到微珠的mRNA分子数(即cDNA分子数)。
Seurat V2的meta.data最初两列:nUMI, nGenes
后续更新的Seurat V3,4的列名分别改为:nCount_RNA,nFeature_RNA
2、cellranger比对#
示例数据:GSE178911共4个样本,每个样本有4个lane的比对结果(SRRxxxxxx id)
(1)原始测序数据fastq.gz
https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/fastq-input
- 如上测序原理的双端测序,一个样本通常会产生一对fastq.gz结果;
- 若同一样本在不同的lane分别测序,则会产生若干对的fastq.gz结果。
如下示例,为样本名为MySample的测序结果。
1
2
3
4
5
6
7
|
PROJECT_FOLDER
|-- MySample_S1_L001_I1_001.fastq.gz
|-- MySample_S1_L001_R1_001.fastq.gz
|-- MySample_S1_L001_R2_001.fastq.gz
|-- MySample_S1_L002_I1_001.fastq.gz
|-- MySample_S1_L002_R1_001.fastq.gz
|-- MySample_S1_L002_R2_001.fastq.gz
|
I1文件表示sample index,用于区分不同样本的标签,在后序的比对分析中可不提供。
在分析公共数据时,可使用aspera或者prefetch方式下载原始数据;并修改成上述的规范文件名。
(2)cellranger比对环境
https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest?
在上述网址内下载比对软件cellranger与相应物种的参考数据集,解压即可使用。
1
2
3
4
5
6
7
8
9
10
|
cellranger-7.1.0/bin/cellranger --version
# cellranger cellranger-7.1.0
tree -L 1 refdata-gex-GRCh38-2020-A/
# refdata-gex-GRCh38-2020-A/
# ├── fasta
# ├── genes
# ├── pickle
# ├── reference.json
# └── star
|
(3)cellranger比对命令
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
bin=/path/to/cellranger-7.1.0/bin/cellranger
ref=/path/to/cellranger-7.1.0/refer/refdata-gex-GRCh38-2020-A
fq_dir=/path/to/PROJECT_FOLDER
$bin count \
--fastqs=${fq_dir} \
--sample=MySample_S1 \
--transcriptome=$ref \
--id=MySample_S1 \
--localcores=10 \
--no-bam \
--nosecondary
# --fastqs 交代测速数据路径(文件夹名)
# --sample 交代待比对样本名的前缀(因为该文件夹内可能有许多样本的测序数据)
# --transcriptome 交代参考基因组文件夹
# --id 交代储存结果的文件夹,如果没有会自动创建
# --localcores 多线程
# --no-bam 不生成bam文件(视情况而定:如果进行RNA速率分析等,需要保留bam文件)
# --nosecondary 不进行后续分析
##输出结果一般在 ${id} 的out文件夹
|
(4)比对结果输出
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
|
tree -L 2 MySample_S1
# MySample_S1
# ├── MySample_S1.mri.tgz
# ├── _cmdline
# ├── _filelist
# ├── _finalstate
# ├── _invocation
# ├── _jobmode
# ├── _log
# ├── _mrosource
# ├── outs
# │ ├── cellsorted_possorted_genome_bam.bam
# │ ├── filtered_feature_bc_matrix
# │ ├── filtered_feature_bc_matrix.h5
# │ ├── metrics_summary.csv
# │ ├── molecule_info.h5
# │ ├── possorted_genome_bam.bam
# │ ├── possorted_genome_bam.bam.bai
# │ ├── raw_feature_bc_matrix
# │ ├── raw_feature_bc_matrix.h5
# │ └── web_summary.html
# ├── _perf
# ├── SC_RNA_COUNTER_CS
# │ ├── CELLRANGER_PREFLIGHT
# │ ├── CELLRANGER_PREFLIGHT_LOCAL
# │ ├── fork0
# │ ├── FULL_COUNT_INPUTS
# │ ├── GET_AGGREGATE_BARCODES_OUT
# │ ├── SC_MULTI_CORE
# │ ├── _STRUCTIFY
# │ └── WRITE_GENE_INDEX
# ├── _sitecheck
# ├── _tags
# ├── _timestamp
# ├── _uuid
# ├── _vdrkill
# └── _versions
|