文献: scGPT: toward building a foundation model for single-cell multi-omics using generative AI
时间:2024 Feb. (Published)
期刊:Nature Method
DOI:https://doi.org/10.1038/s41592-024-02201-0
Github:https://github.com/bowang-lab/scGPT
1. 简介
1.1 关于作者
通讯作者:Bo Wang,多伦多大学
- https://wanglab.ai/
- https://github.com/bowang-lab/
- https://scholar.google.com/citations?hl=en&user=37FDILIAAAAJ

1.2 文献概述
-
受启发于GPT等基于transformer的大模型,Wang团队基于大量单细胞转录组数据,构建了scGPT foundation model,可用于多种常见的单细胞下游分析任务,性能优于其它已有的分析工具。
-
核心假设:
- 在NLP领域中,一个句子由多个单词组成。经过预训练的自监督学习,foundation model可以提取输入句子中每个单词(或者句子整体)的高级表示(Embedding);
- 而在scRNA-seq中,一个细胞可以认为由所有基因的不同程度表达定义。经过预训练的自监督学习,foundation model可以提取输入单细胞表达数据的每个基因(或者细胞整体)的高级表示(Embedding)。
-
在Pre-trained foundation models基础上,将提取的基因/细胞Embedding在具体的单细胞下游分析任务中进行二次微调(fine-tune),发挥大数据生成的强大优势。
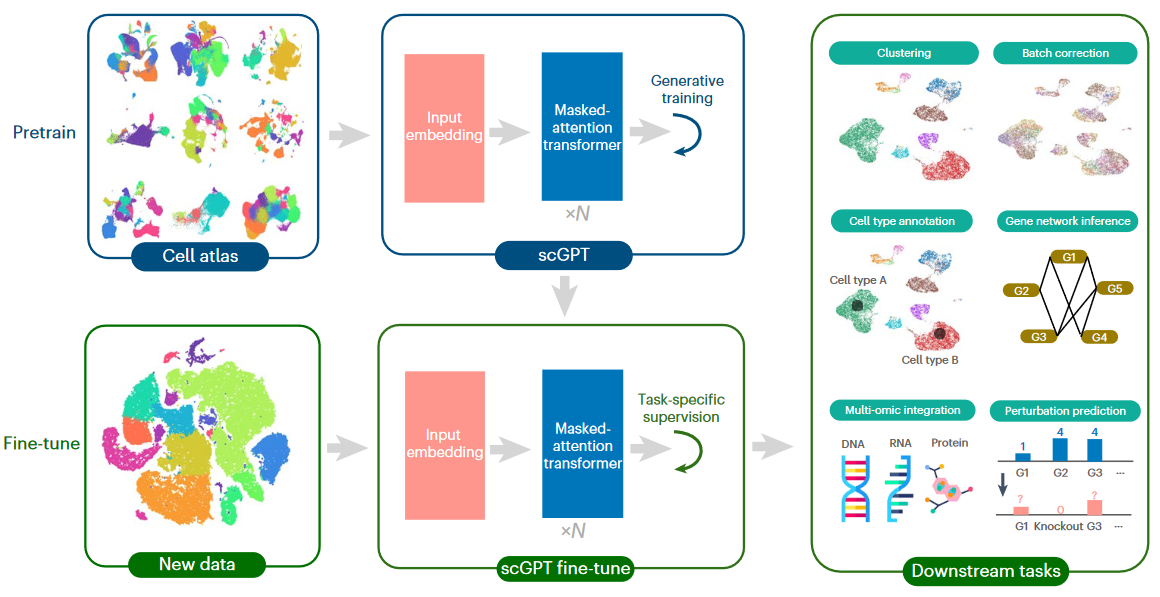
2. Pretrain预训练
Pre-train foundation model的核心功能就是以一个单细胞的数据为输入,将提取的Gene/Cell Embedding为输出。
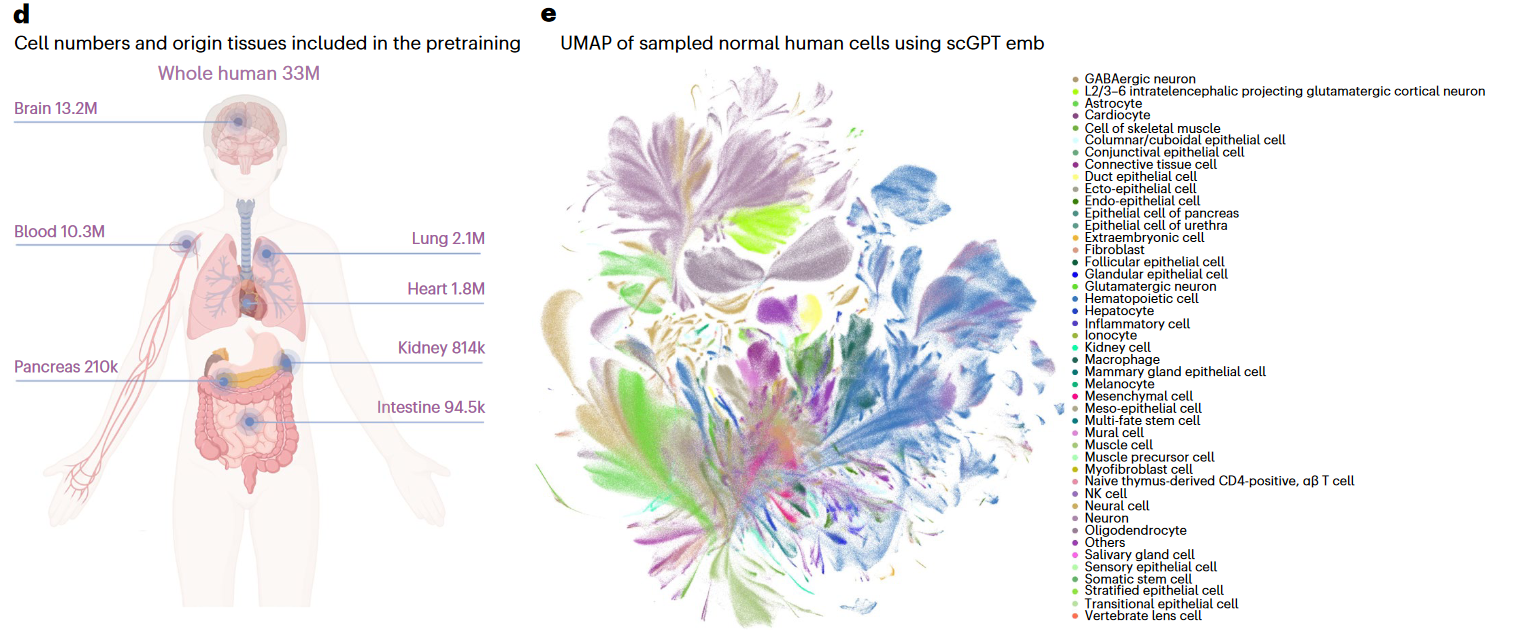
2.0 数据规模
- 文章从CELLxGENE网站收集了33M个正常细胞的scRNA-seq数据用于训练Foundation model
- 其中来自Brain、Blood等器官的细胞数最多
2.1 初始化输入
- scGPT的原始输入数据通常为N个细胞,G个基因的Count表达矩阵。此时,需要对每个细胞的基因数据进行初始化处理,作为scGPT的标准输入;
- 概括来说,分别从每个基因的三个角度进行D维嵌入编码 (1×D),然后再进行矩阵加法(仍为1×D),最终将所有基因合并得到(M×D)的细胞特征矩阵。
因为细胞的基因会根据情况进行选择,并且考虑到<cls>等特殊词元,所以这里并不是G×D
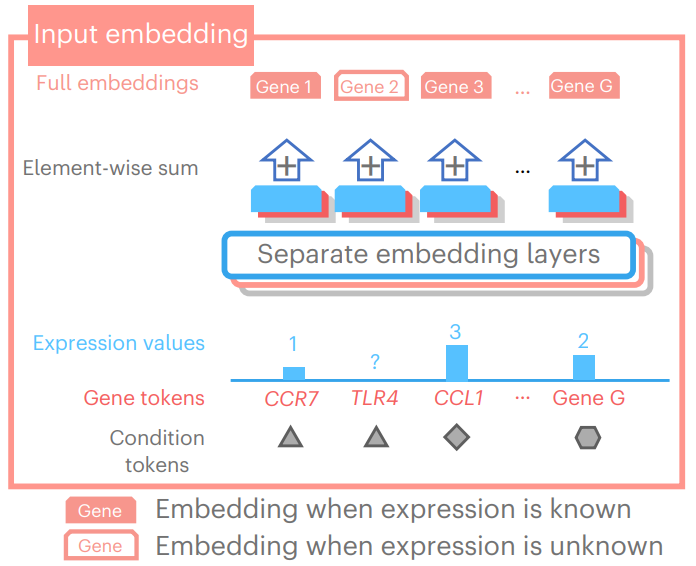
(1)Gene token
- 所有基因(词元)构成的词表中,每个基因都有一个对应的整数标识符(Integer Identifier)
- 此外还有其它特殊的词元,例如<cls>, <pad>等
- <cls>词元通常放在细胞M个词元中的第一个,不表示特定基因,而用于cell representation
- <pad>词元用于补长至固定长度M(e.g. 有些情况不考虑表达值为0的基因时)
- 采用PyTorch embedding layer将投射成D维的Embedding
(2)Gene expression
- 单细胞的count表达值在不同测序背景的条件下,不具有可比性。
- scGPT采用bin分段处理,将单个细胞i的非零基因表达值,分为k个bins,分数分别是1..k。
- 例如,若基因bin打分为k,表示其表达值处于最高的bin范围内;
- 每个细胞分bin的阈值标准都会不尽相同。
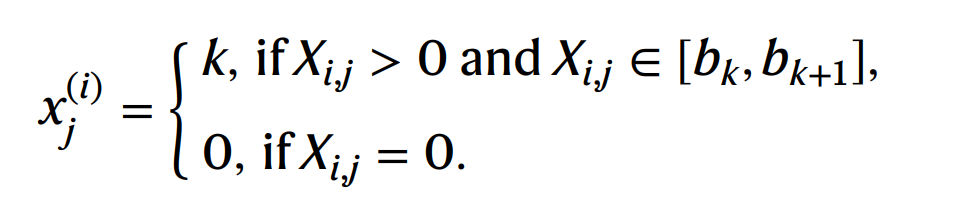
- 采用MLP,转换为D维的Embedding
(3)Condition token
- 记录基因额外的Condition token,例如是否为perturbated genes
- 采用PyTorch embedding layer将投射成D维的Embedding
综上,一个细胞(i)的初始化输入h (M × D)的计算方式如下图所示。其中第一列通常表示为<cls>作为Cell representation;其它列(除<pad>等特殊字符外,均表示Gene Embedding。

2.2 自监督训练
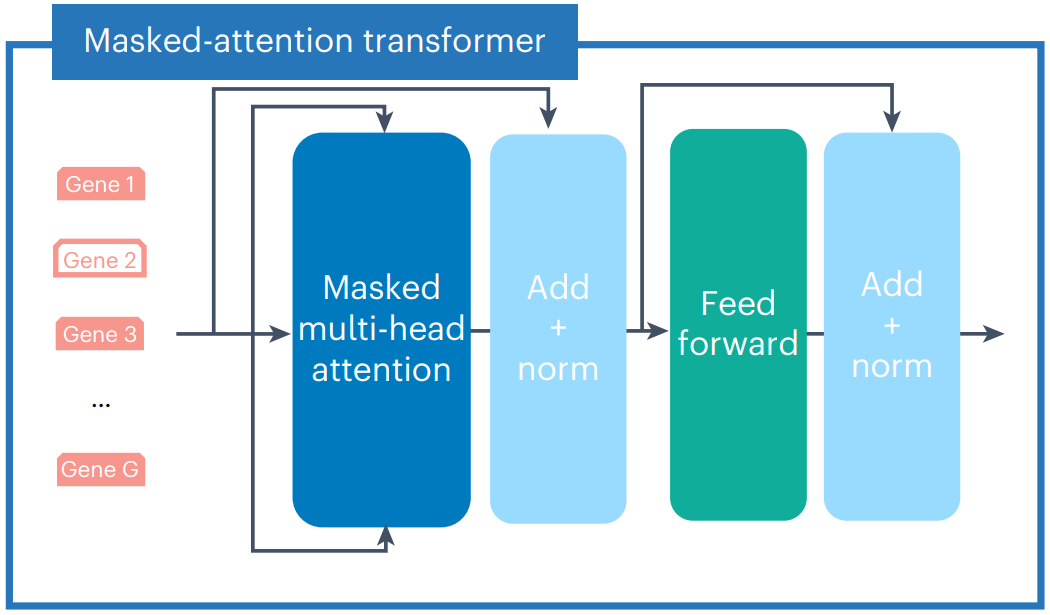
(1)Transformer块
- Foundation model主要由 12 (
l)个Transformer块组成,每个块则均采用了多头(8)自注意力机制;

-
由于这里的M通常较大,即一个细胞考虑数千上万个基因(可以理解为特别长的句子),scGPT采用了FlashAttention算法用以加速自注意力计算。
此外,对于每个细胞,只有表达值非0的基因参与预训练过程,以提高速度。
-
作者也推荐了其它高效计算的Transformer变体,包括linear complexity (Linformer),Kernelized Self-Attention (KSA)。
(2)自监督任务
- scGPT自监督任务的核心是预测掩码(masked)基因的表达值水平,采用MSE损失函数。
- 具体分为如下两个子任务:
- Gene-prompt:基于已知表达值的Gene Embedding,预测未知表达值Gene的expression value;
- Cell-prompt:基于<cls> cell representation预测全基因的expression value。
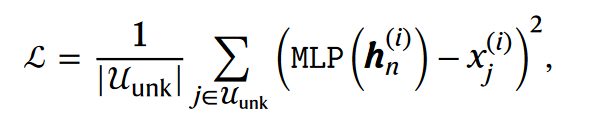
- 然后,将上述两种模式的损失loss相加后,再计算梯度并更新模型参数。
(3)Masked Attention
- 自注意计算过程为:将masked gene Embedding( without expression) 作为query,计算与其它known gene Embedding(也包括自己)的注意力权重后,再进行加权运算得到输出;
- 与GPT模型所处理的文本句不同之处在于:the non-sequential nature of the genes in one cell. 为此作者设计了masked attention注意力计算方式;
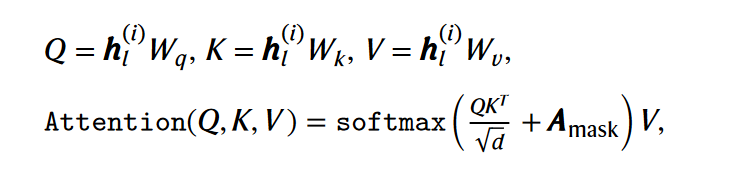
- 如上公式,在不考虑Amask的情况下,为标准的自注意力计算方式。而Amask可参考如下公式,以及下图左A(行表示query,列表示key)理解。每一个query(i)计算与其他词元注意力(包括与它自己)时:
- 若key (j)不是unknown expression gene,则为0;
- 若i = j时,且j是unknown expression gene,则为0(自己与自己的注意力计算)
- 其它情况下,则为负无穷(对应图中的深蓝色单元格)
- 每一行(i)表示一个query词元与该细胞所有词元的注意力计算。
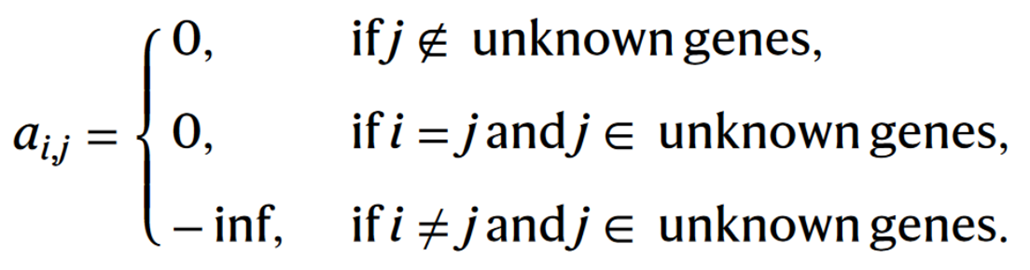
- a = 0表示不产生任何影响,a = -inf表示将query(i)对于key(j)的注意力置换为-inf,经softmax转换后则变为0。
The rule of thumb for scGPT attention masking is to only allow attention computation between embeddings of the ‘known genes’ and the query gene itself.
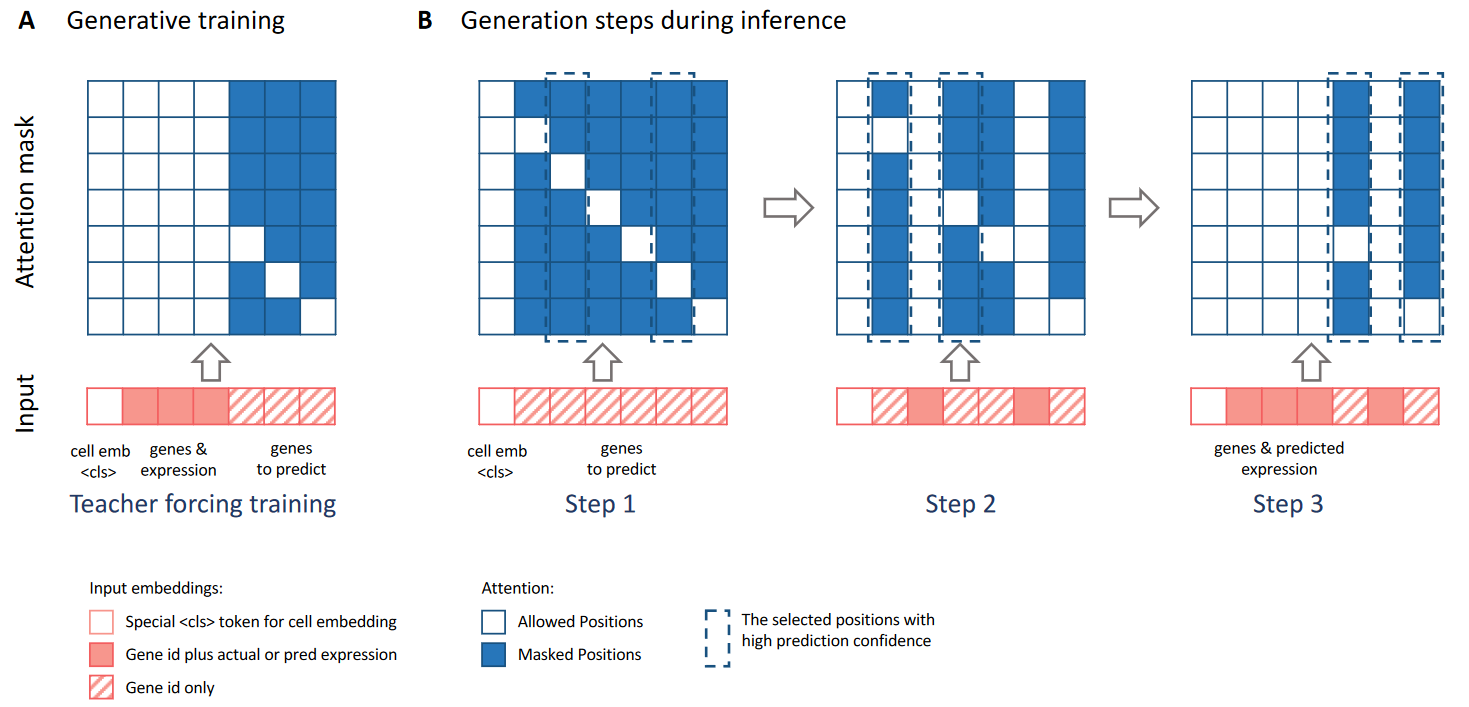
- 在一个cell的全部masked gene表达值预测的过程中,采用多轮(k)迭代预测的思路(如上图右,类比GPT的自回归训练)
- 在第一轮中,对于所有masked token的预测,将其中1/k个high prediction confidence的token标注为known genes。
- 然后在新一轮迭代中,重复上一步骤,直至预测出所有的masked token。
- 值得注意的是,文章并没有说明prediction confidence是如何计算的。个人理解本质还是基于与真实表达值之间的误差。
In each generation iteration, scGPT predicts the gene expression values of a new set of genes, and these genes in turn become the ‘known genes’ in the next iteration for attention computation.
2.3 多批次与多模态表示
- 在多批次、多组学合并下游任务中,需要额外的token Embedding以表示必要的批次和组学信息,供模型学习;
- 但是scGPT在Transformer的pre-train过程中,并未加入相关信息。而是在foundation model的输出结果中再进行拼接操作,即模型在Pre-train后,Fine-tune前引入批次和模态信息,以希望在微调过程中学习到相关信息。
- 这样做的主要原因是:如果批次和模态信息在输入阶段被引入,Transformer的自注意力机制可能会过度关注同一模态或批次内的特征,导致模型忽略跨模态或批次的重要关联。
This is to prevent the transformer from amplifying the attention within features of the same modalities while underestimating those of different modalities.
- modality tokens (tm):表示词元对应的feature是Gene/Protein/Peak中的哪一种;
- batch tokens (tb):表示token是否来自一个批次,通常对于细胞来说的。因此一个细胞的batch token都是一样的。
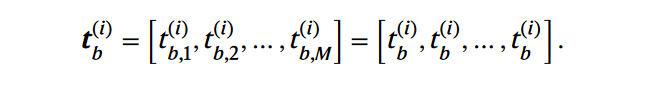
- 如下公式,表示同时存在多批次以及多组学的情况
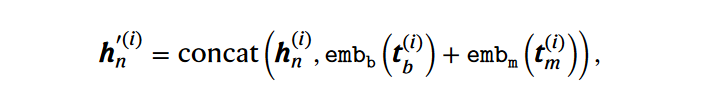
- 如下公式,表示同种组学,存在多批次的情况
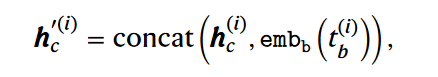
3. Fine-tune微调
3.1 细胞类型注释
(1)Method
-
对于Foundation model提取的每个细胞Cell Embedding (<cls>) 构建一个MLP分类器,用以预测细胞的类别,并使用交叉熵作为损失函数。
-
首先使用一个标注细胞类型的数据集(reference set)进行Fine-tune,然后再使用一个Held-out数据集验证
-
输入数据前的预处理:
- common set between foundation model and the input data
- Gene expression: normalization → log1 → bin
- All gene as input (include zero expression)
(2)Result
-
在Human pancreas (胰腺) dataset
- 对于每种细胞的预测Precision都达到0.8以上 (预测为该细胞类型中,实际为该细胞类型的比例)
-
以Human immune cells进行fine-tune,预测多发性硬化症(MS)的细胞类型
- 平均准确率Accuracy可以达到0.85左右
-
使用6种肿瘤细胞类型作为fine-tune,预测3种其它肿瘤的细胞类型
- 在肿瘤微环境细胞类型预测同样表现良好
-
与TOSICA、scBERT模型进行了比较,均表现出一定的优势
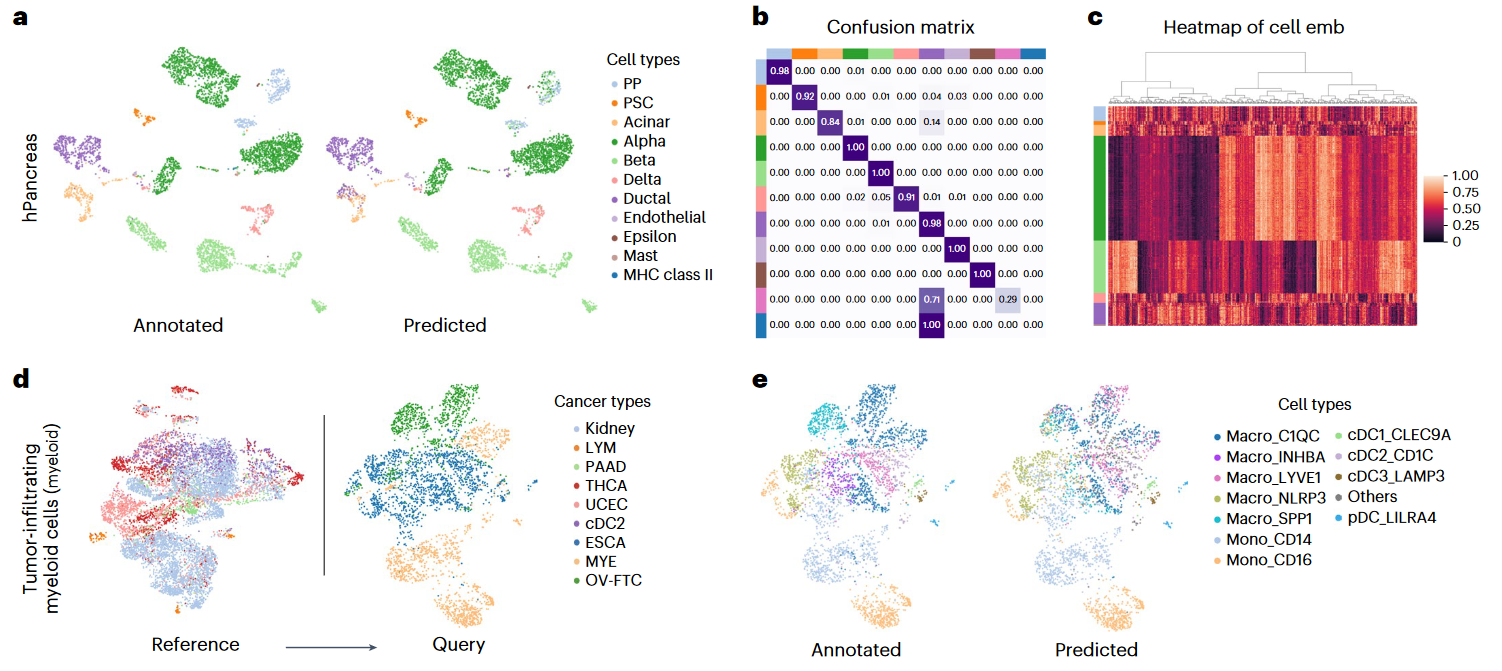
此外,作者还提出了Reference mapping注释方法:在Zero/Fine-tune模型时,获得标签细胞类型的Cell Embedding。然后再计算出Query cells的Embedding。最后据此,计算出每个Query cell最相近的Reference cells,从而注释其细胞类型(KNN)。
3.2 基因扰动预测
(1)Method
- 对于Foundation model提取的每个细胞的Gene Embedding,将一部分基因的表达值掩码,从而进行Fine-tune微调训练,并以MSE作为损失函数;
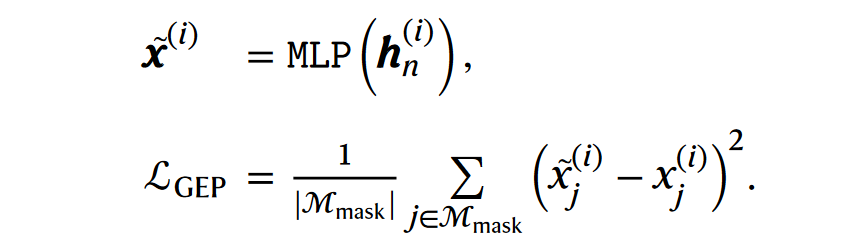
- 在Perturbation prediction task:
- Input: Gene expression为Control cell expression, Condition token标注相应位置Gene是否被Perturbate
- Output: Post-perturbation expression
- 输入数据前的预处理:
- only select HVGs for training
- Log1p expression instead of binned values (output相同)
(2)Result
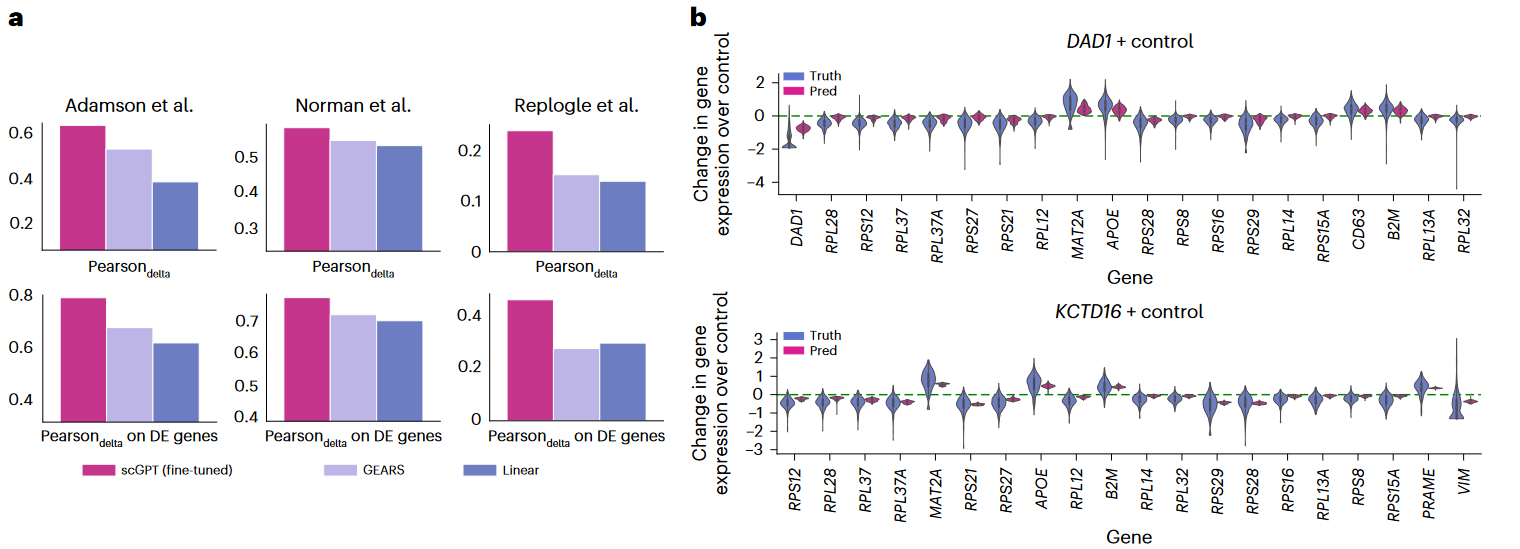
- 分别基于三个单细胞CRISPR数据集,比较了scGPT与Linear、GEARS模型的性能表现
- Adamson: 87 one-gene perturbations;
- Replogle: 2823 one-gene perturbations;
- Norman: 131 two-gene and 105 one-gene perturbations
- 对于每个数据集,使用一部分的扰动数据进行fine-tune,再对其余unseen gene的扰动数据作为test
- 评价指标为计算预测与真实的post-perturbation expression的相关性 (基于全部基因,或者是前20个影响最显著的基因)
- 结果发现scGPT模型相比于其它两种模型,提高了5–20%
(3)In silicon reverse perturbation prediction
- 简单理解,根据perturbation expression的结果反向预测是最有可能哪个基因被扰动;
- 参考作者在Github issue (https://github.com/bowang-lab/scGPT/issues/87)的解答:
- 其Fine-tune步骤其实与上面Perturbation prediction task基本一致;
- 使用数据集的一部分进行微调后,再预测所有相关基因扰动的表达结果;
- 最后使用真实的Query perturbation上述的预测结果进行KNN关联分析。
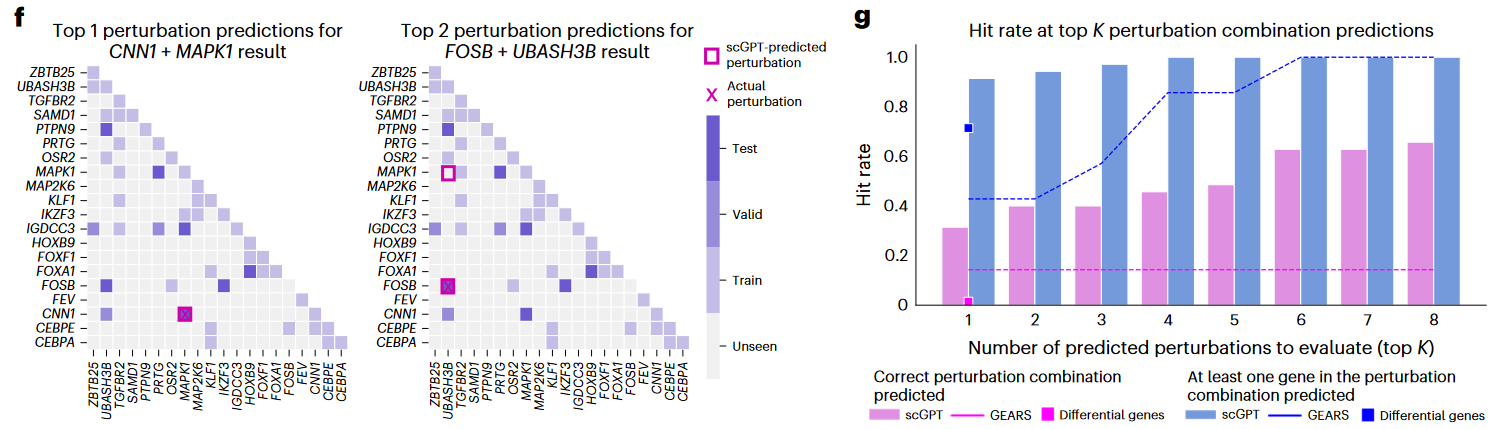
The model was fine-tuned in the same way as in the “forward” perturbation prediction. It used a subset of the dataset as we illustrated in Figure 3F. The reverse perturbation task utilized the model in a different way. To summarize, the result cell states of all possible perturbations were predicted by the fine-tuned model, and then an actual sequenced cell state can query all the predicted cell states in a nearest neighbor search manner, so that the retrieved neighbors indicate the possible origin perturbations.
3.3 多批次/组学整合
(1)Method(Multi-batch)
-
核心目标:对于多批次的scRNA-seq数据,优化不同批次中每个细胞的cell representation
- correct batch effects while preserving biological variance
-
输入数据的预处理
-
Common set between foundation model and the input data
-
Gene expression: normalization–log1–bin
-
All genes as input (include zero expression)
-
如2.3中所述,在微调前,还需要在Foundation model的输出结果中,补充细胞的批次信息。
-
-
具体在Fine-tune训练中,设计了对多个目标函数进行损失计算,以共同用于模型优化
1)GEP:基因表达预测,参考3.2;
2)GEPC, Gene expression prediction for cell modeling,即为了优化Cell Embedding(hc),而预测基因表达。
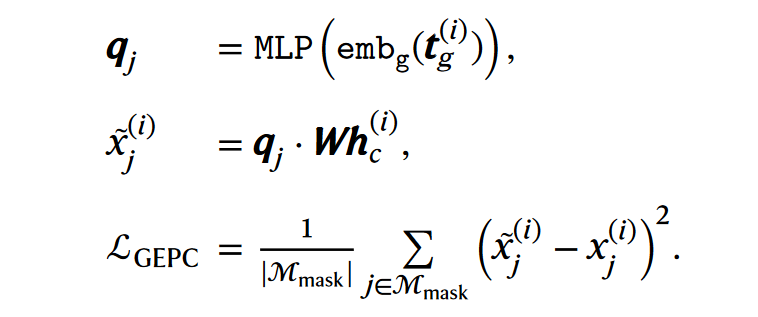
3)ECS, Elastic cell similarity:基于一个mini-batch的两个细胞的相似度计算损失函数。使得高于某个阈值的两个细胞更相似,低于的则更远离。
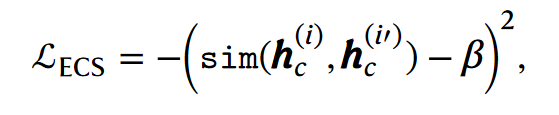
4)Domain adaptation via reverse back propagation:主要作用是使得生成的cell representation表示经过MLP分类器无法预测其正确的batch信息(类似于GAN 对抗神经网络)。本质上还是建立MLP预测Batch的分类器,但是计算梯度后,进行反向更新参数。
(2)Result
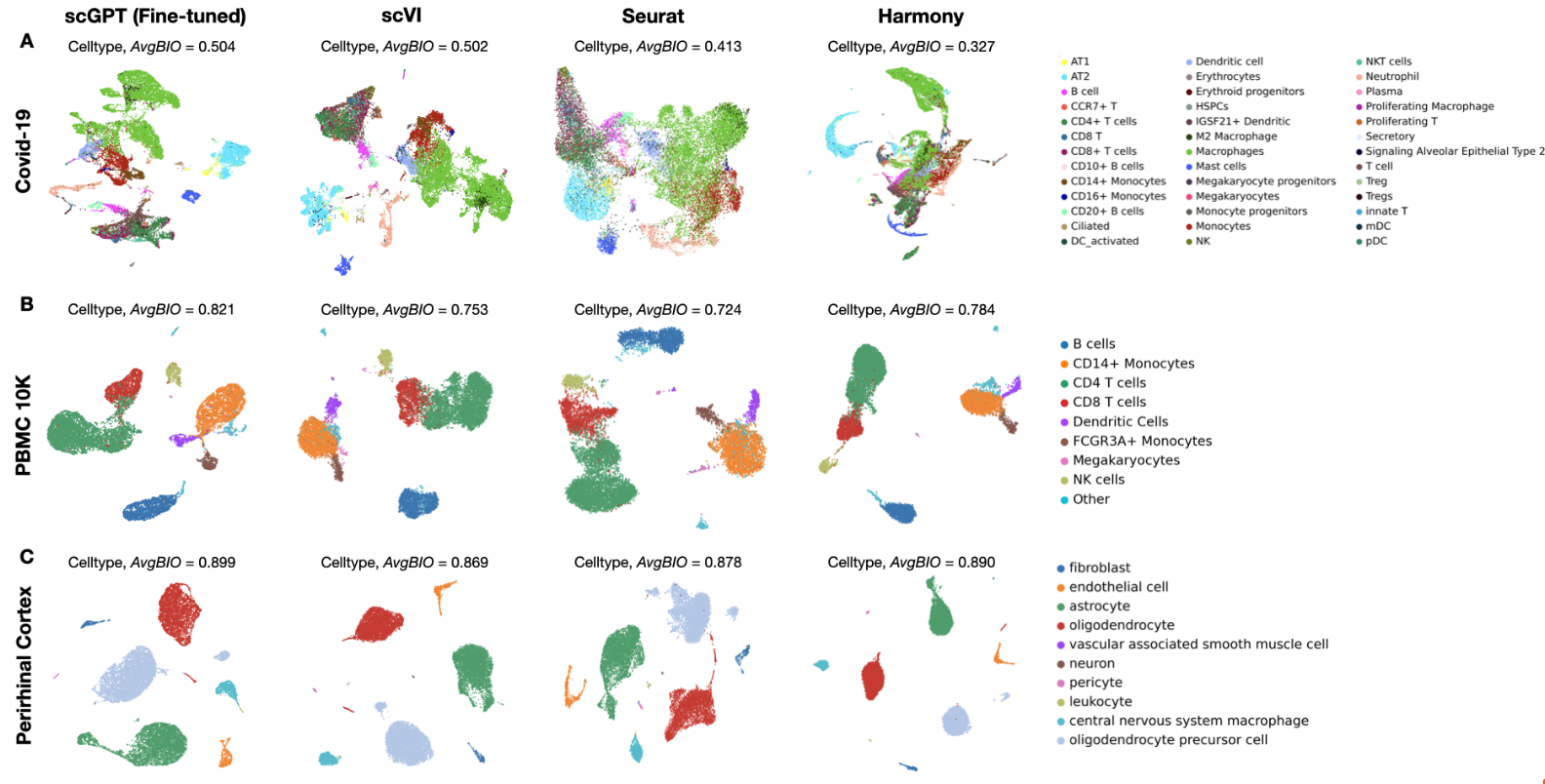
-
基于三个数据集,比较了scGPT与scVI,Seurat,Harmony三个工具去批次的效果
-
COVID-19: 18 batches
-
PBMC-10K: 2 batches
-
Perirhinal cortex: 2 batches
-
-
评价指标:基于三个指标(NMI/ARI/ASW)的AvgBIO,值越高表明去批次效果越好。
- scGPT均优于其它三种工具的效果。
(3)Multi-omics integration
Method
- 目标是使得相同细胞类型的不同组学数据能够有相似的Cell Embedding,从而在聚类时比较接近。
- scGPT主要考虑了3种组学,分别是scRNA-seq、scATAC-seq,Single-cell proteomics。对此,有两种数据形式:
- paired setting:一群细胞同时测多种组学
- mosaic setting:一群细胞测一种组学,另一群测另一组。
- 对于scRNA-seq可以直接继承第2节训练的Foundation model,对于其它两组需要重头训练。
- 在Pre-train之后,如2.3小节需要拼接表示多模态的token(如果涉及多批次,也要添加batch token)
- Fine-tune 目标函数包括GEP,GEPC(如果涉及多批次,还需要添加DAR)
Result
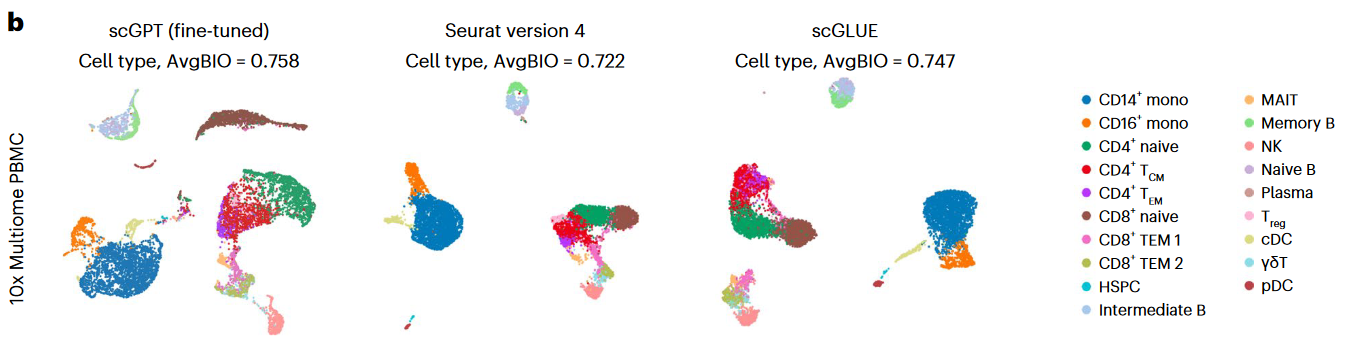
-
对于Multiome PBMC (paired)数据集,与scGLUE,Seurat进行了比较,如下图所示
- RNA-seq、ATAC
-
对于Bone marrow mononuclear cells (paired)数据集,与Seurat进行了比较
- RNA-seq、Protein
- 9w个细胞,12个donor(multiple batches),48种细胞类型
-
对于ASAP human PBMC (mosaic)数据集,与scMoMat进行了比较
- 4个批次,3种组学
3.4 基因调控网络构建
(1)Method
-
本质上是基于Foundation model或者Fine-tune model提取的Gene Embedding计算两两基因间的相关性,用来构建gene similarity network。
-
Zero-shot setting模式下,直接使用Foundation model输出的Gene Embedding计算;
-
Fine-tuned setting模式下,使用特定数据集微调后输出的Gene Embedding计算
使用integration task进行微调,以学习特定数据集相关的Gene Embedding (下同)
-
-
基于注意力机制的target gene鉴定:Attention map可以反映出基因之间的相互影响。其中,每一列(column),表示这个gene(列名)对所有query gene的influence。
-
使用perturbation datasets可以帮助推测perturbating gene的target,如下图所示
- 首先,基于control cell得到的attention map;
- 然后,基于perturbation expression得到的attention map;
- 计算上述二者对的差值,可以推测特定基因干扰前后,影响最大的gene
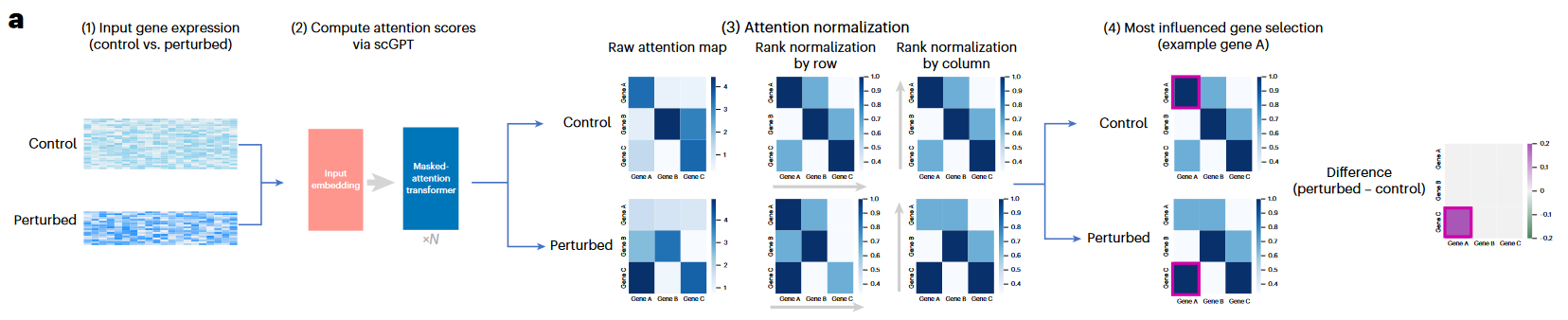
(2)Result
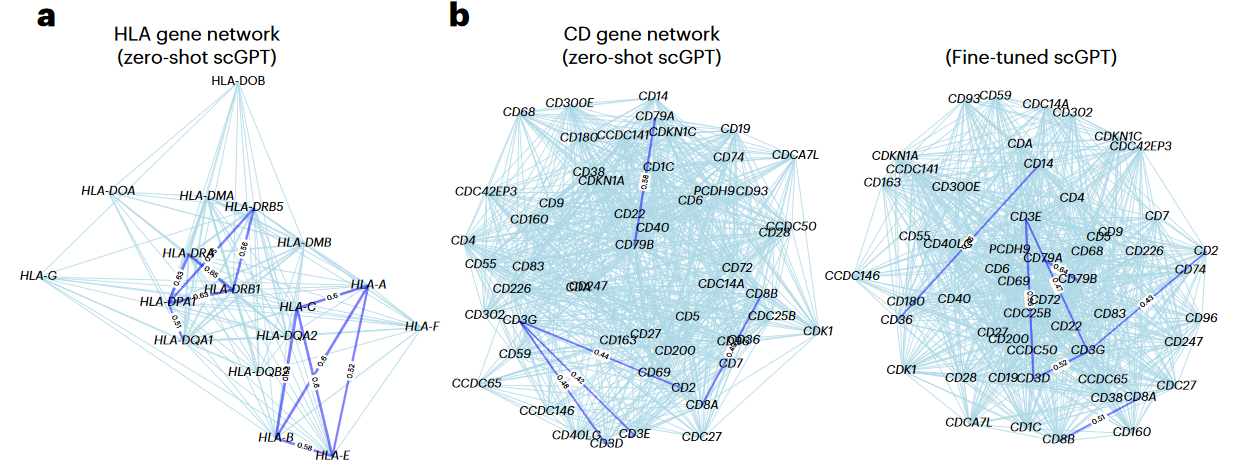
- 文章首先对zero-shot模式进行了探索,然后在对一个human immune dataset进行了Fine-tune后构建GRN,均发现了具有生物学意义的调控网络以及Gene cluster (Leiden),并进行了通路富集分析等。
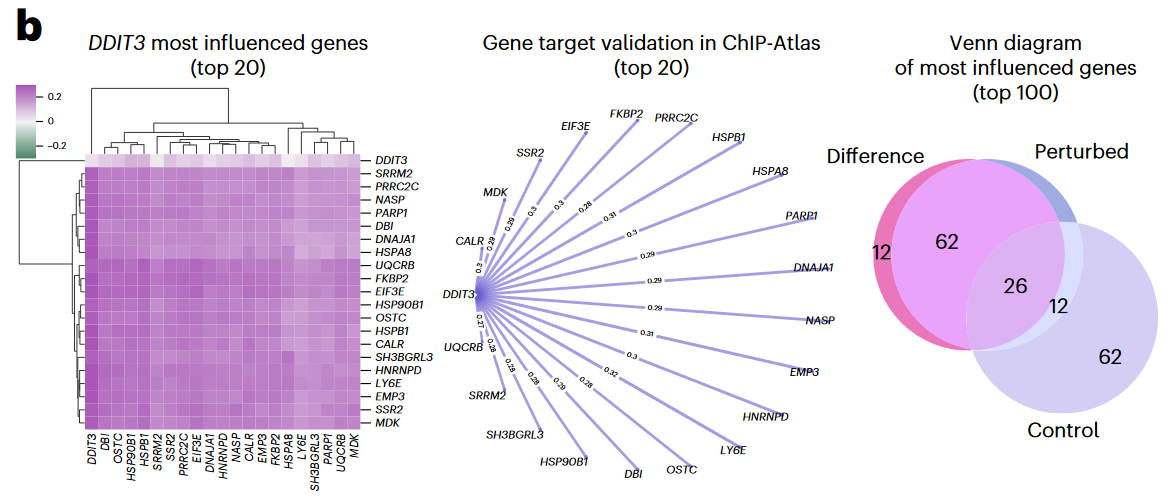
- 接下来,作者基于Pre-train scGPT blood model,使用了Adamson CRISPR数据集(87 CRISPR inference on leukemia cells)进行了微调。
- 例如下图,通过比较DDIT3扰动前后的difference attention score, 鉴定了其影响最大的的Top20/100个基因。
4. 模型影响因素
- 综上,scGPT Foundation model在处理下游分析任务时,相比于其它已有的单细胞工具均表现出明显的优势;
- 最后,文章讨论了两个可能影响Pre-train model性能表现的因素。
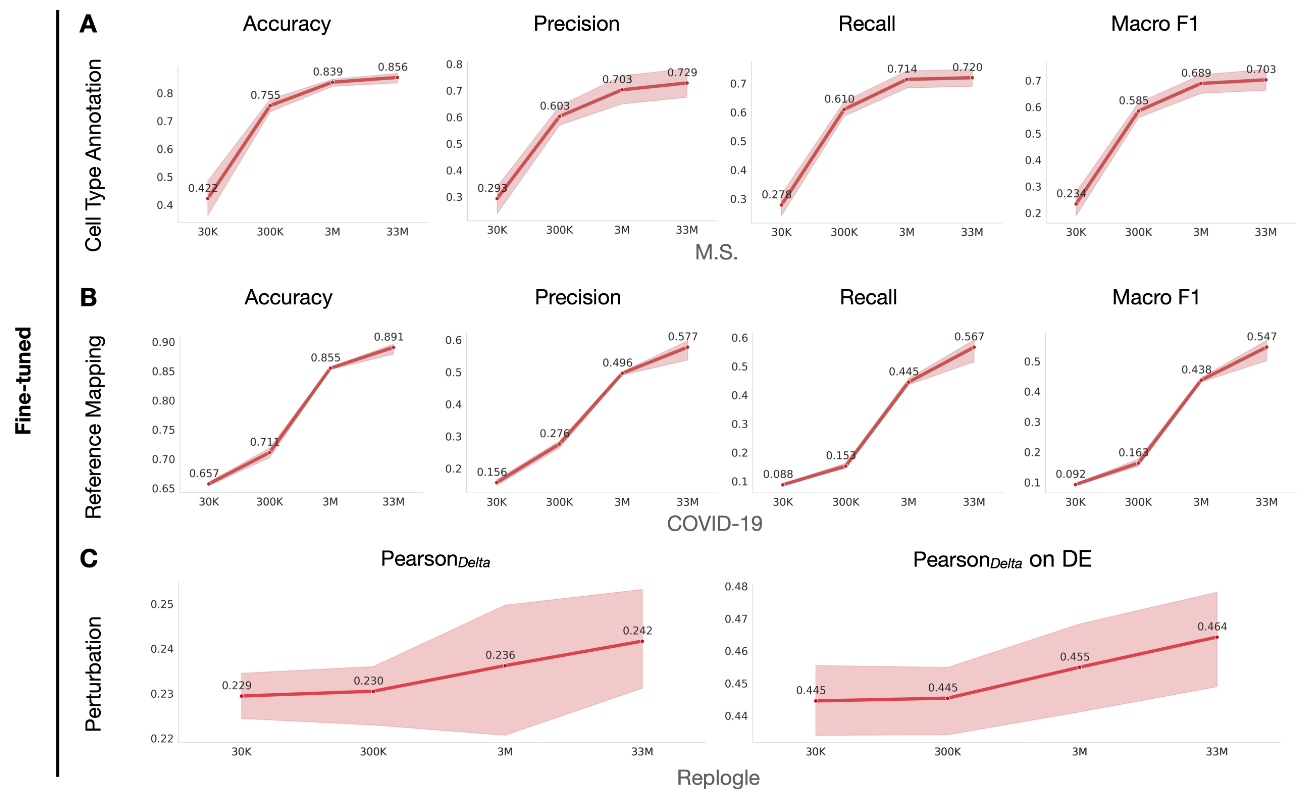
4.1 训练样本量
- 首先,文章研究了训练样本量对于预训练模型的影响;
- 如下图,结果发现数据量最多时(从30K到33M),模型在多种下游任务中的表现越好;
- As larger and more diverse datasets become available, we can anticipate further improvements in model performance, advancing our understanding of cellular processes.
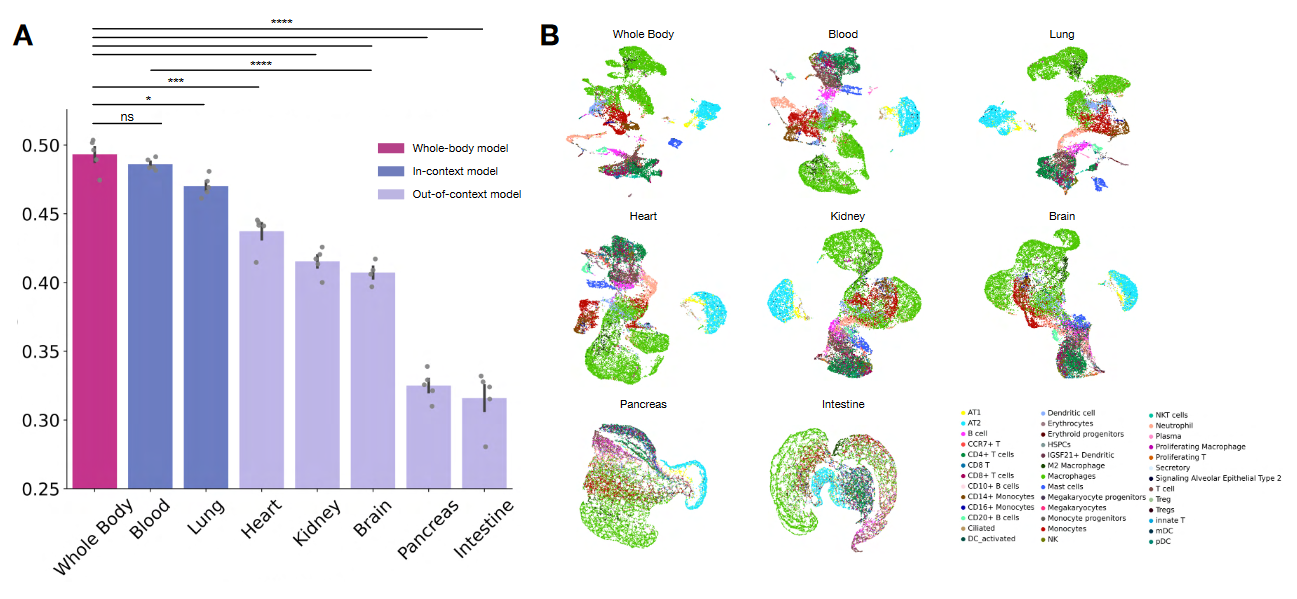
4.2 训练样本类型
- 文章进一步研究了细胞来源对于模型的影响;
- 在COVID-19数据集的多批次合并任务中,结果发现:
- 使用全部33M数据与仅使用Blood数据的Foundation model的性能比较接近;
- Lung来源的细胞量尽管只有2.1M,远少于Brain细胞量(13.2M),但前者性能明显表现更优。
- This emphasizes the importance of aligning the cellular context in pretraining with the target dataset for superior results in downstream tasks.
后续将参考scGPT工具的教程手册及源代码,学习其构建细节与数据处理方式等。