题目:Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance
期刊|日期:nature medicine | 26 April 2023
DOI:https://doi.org/10.1038/s41591-023-02371-y
简介:来自德克萨斯大学安德森癌症中心的Linghua Wang研究团队收集了16种肿瘤类型的>30W个T细胞表达数据,对多种T细胞亚型及其分子、临床特征展开了深入地分析。值得一提的是文章发现了一种曾被忽视的Tstr亚型,并揭示了其与免疫治疗抵抗的关系。
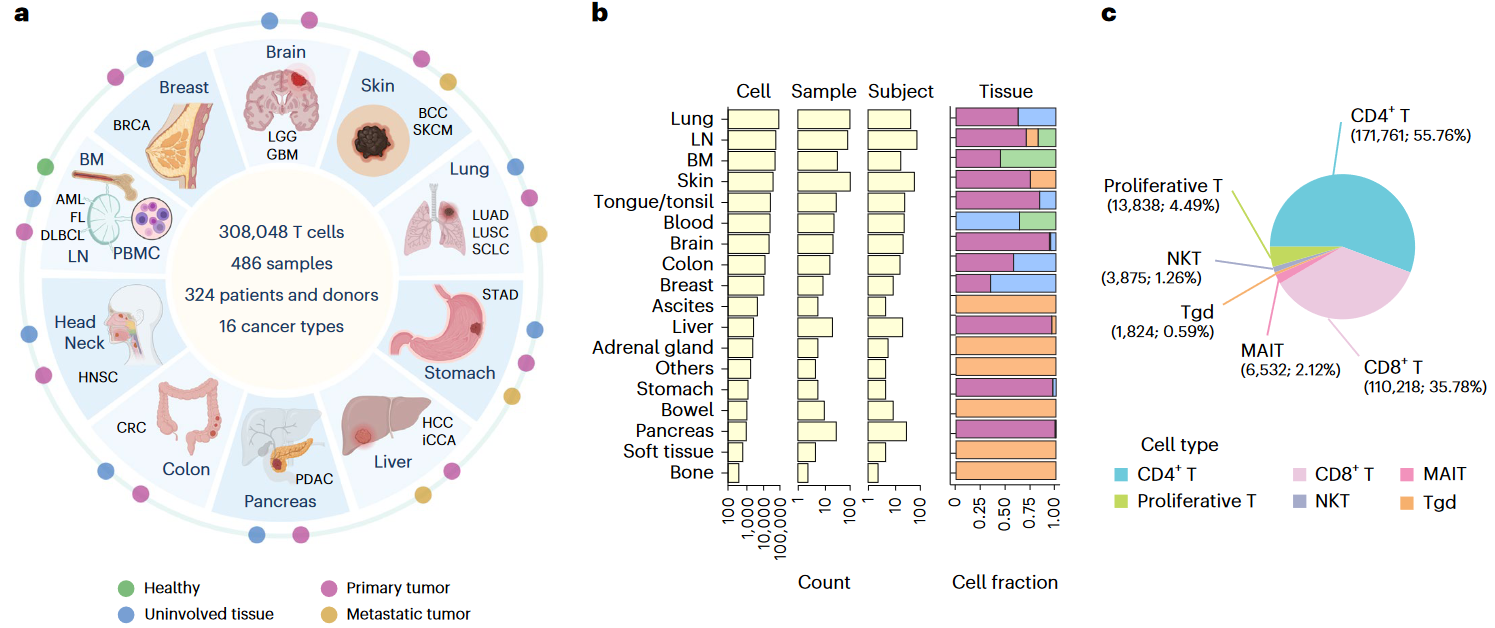
1、数据整体概况
(1)数据规模
- 27个单细胞数据集(10个为内部测序数据,其余17个来自公共数据),涉及16种癌症
- 测序样本根据病理情况可分为healthy, 癌旁的uninvolved, primary以及metastatic tumor
(2)初步分析
- 经合并、过滤处理(method部分有详细介绍)后,共得到308,048个T细胞表达数据
- 初步分群注释后共得到6种主要的T细胞类型
- CD4+, CD8+, γδ T, NKT, MAIT,以及proliferative T cells
- 其中CD4+, CD8+占比最多,分别达55.76%与35.78%
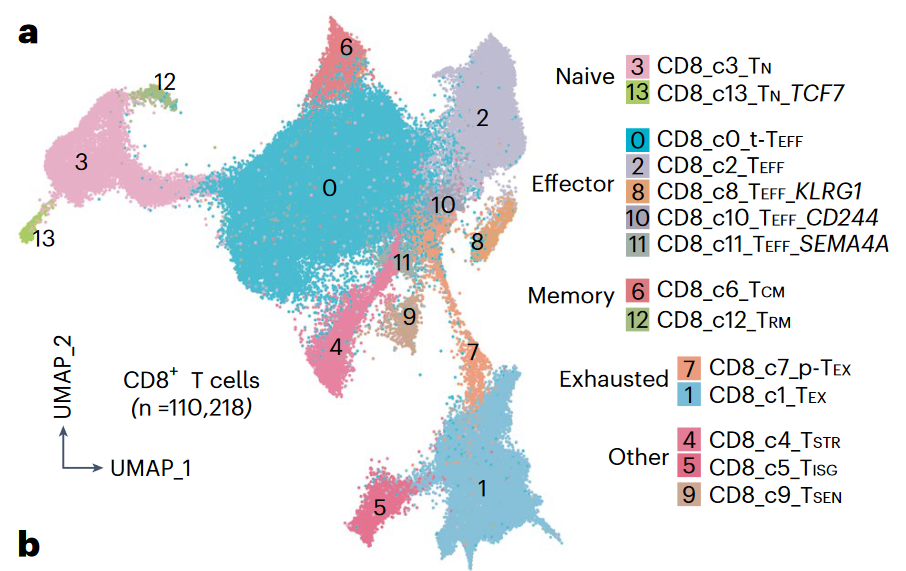
2、T细胞亚型注释
(1)CD8+ T细胞亚型分析
-
亚群分析得到14个 clusters/states,其中可分为Naive,Effector,Memory, Exhausted以及其它;
-
在其它类中的c4_Tstr特异性高表达应激相关的热休克基因(例如HSPA1A和HSPA1B);
-
在拟时序分析中,鉴定出以Naive为发育起点的3条发育轨迹,其中一条的终点是Tstr;
-
进一步观察了特定亚型在不同样本病理状态下的比例分布差异。
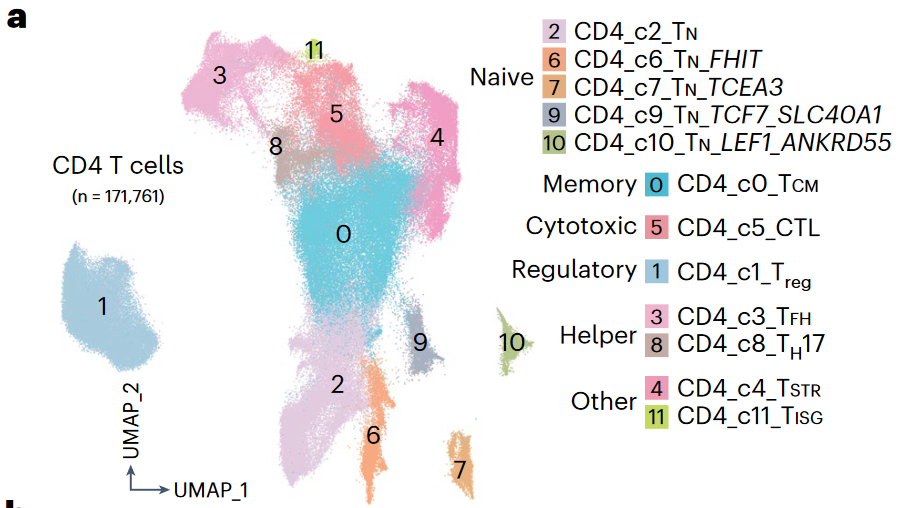
(2)CD4+ T细胞亚型分析
- 群分析得到12个 clusters/states,其中可分为Naive,Memory,Cytotoxic, Regulatory,Helper以及其它(也包括一类Tstr);
- 同样观察了特定亚型在不同样本病理状态下的比例分布差异;
- 考虑到Treg与Tfh类亚型的异质性,进一步对这二者进行亚群分析,分别得到7、5个cluster。
(3)其它类型T细胞
- 最后同样对γδ T, NKT, MAIT,以及proliferative T cells进行亚型注释以及分析。
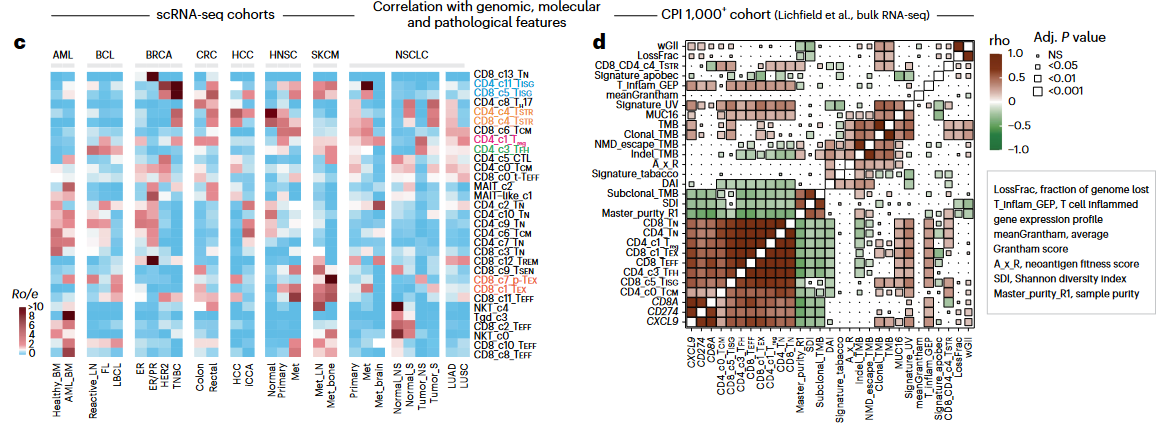
3、亚型特征相关分析
(1)T细胞亚型间相关性
- 使用层次聚类,将上述31种T细胞state可分为4大类;
- 基于样本水平亚型比例相关性,分析存在正相关或负相关的 state-pairs
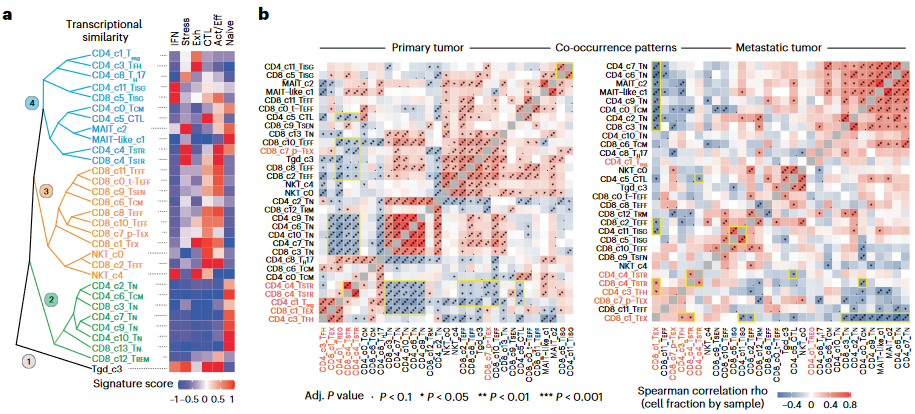
(2)T细胞亚型与样本表型相关性
- 使用特定肿瘤的scRNA-seq数据集,分析特定亚型在normal/primariy/metastase状态下的分布差异;
- 使用TCGA预后数据以及特定亚型的signature,分析生存相关的亚型;
- 使用CPI1000数据集,分析与突变、ICB治疗相关的亚型。
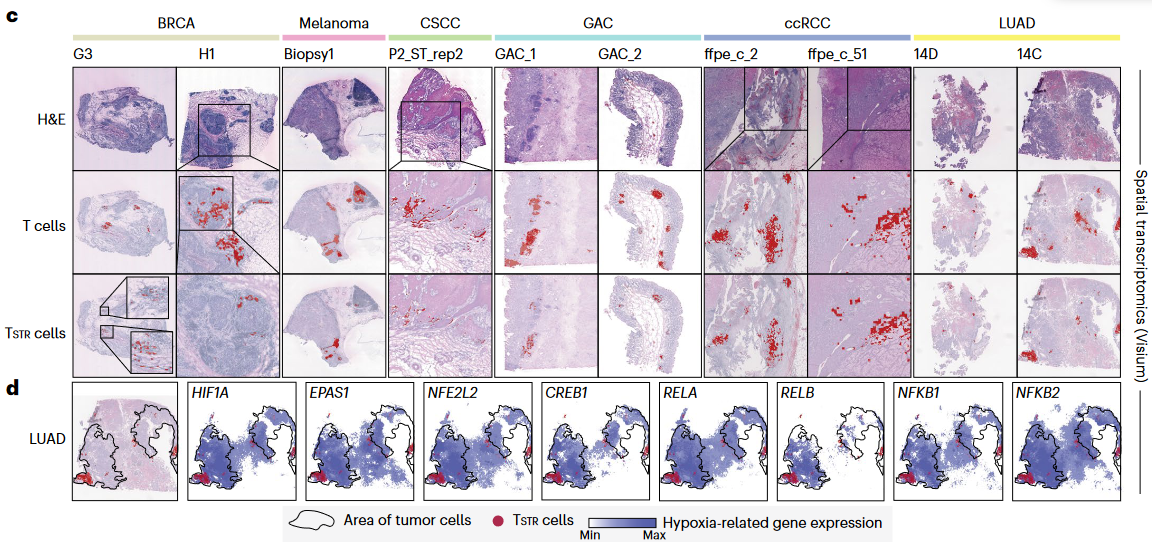
4、Tstr亚型临床意义
(1)验证Tstr亚型的存在,而非细胞裂解时的人为刺激因素
- 通过黑色素瘤的situ hybridization验证HSPA1B的表达
- 通过MSCLC的空间分子成像验证HSPA1A,HSPA1B的表达
- 通过多种癌症的空间转录组学验证HSPA1A,HSPA1B的表达
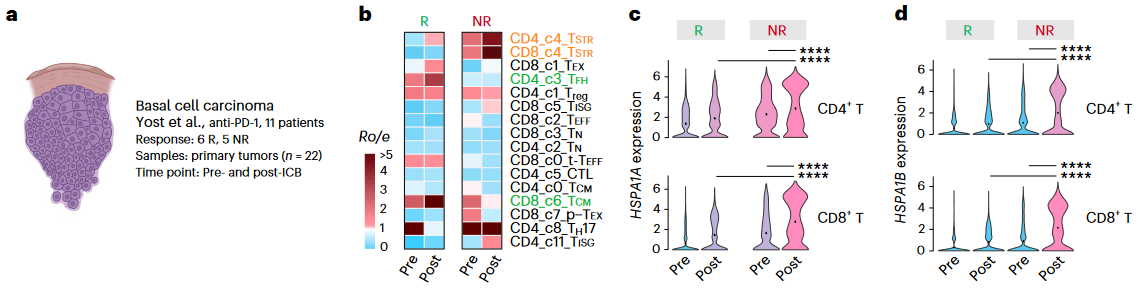
(2)Tstr亚型与免疫治疗的关系
- 另外收集了6个肿瘤scRNA-seq数据,涉及到经anti-PD-1/PD-L1治疗有或者没有响应的样本;
- 观察发现在Tstr均相对富集在non-response的样本组中,提示其与ICB resistance可能有关。
小结:
**(1)**文章本身对于肿瘤T细胞方面深入探索的意义是一方面,其对特定细胞类型在肿瘤(其它疾病)中的多角度分析思路也可以给我们提供一些参考价值。
**(2)**这篇文章在前期的数据收集与前期处理方面下了一番功夫。虽然作者提及将数据整理成了一个网页(https://singlecell.mdanderson.org/TCM/) ,但好像并不提供数据下载。在原文中也仅是提供了公共数据来源(部分还需要申请),所以想直接使用文章数据有一定门槛;
**(3)**在method方法部分,文章较为详细交代了数据前期处理的方法(Seurat),可供借鉴。文章提到对harmony与Seurat-rPCA两种批次整合方法进行了比较,结合silhouette score轮廓分数治疗最终选择了后者。
**(4)**最后作者也在Github上提供了部分绘图R语言代码(https://github.com/Coolgenome/TCM),如对文章某一张图感兴趣,可以学习一下。例如其在比较细胞亚型在不同分组样本的分布差异时,绘制了一种特殊的DimPlot点图。如下所示。
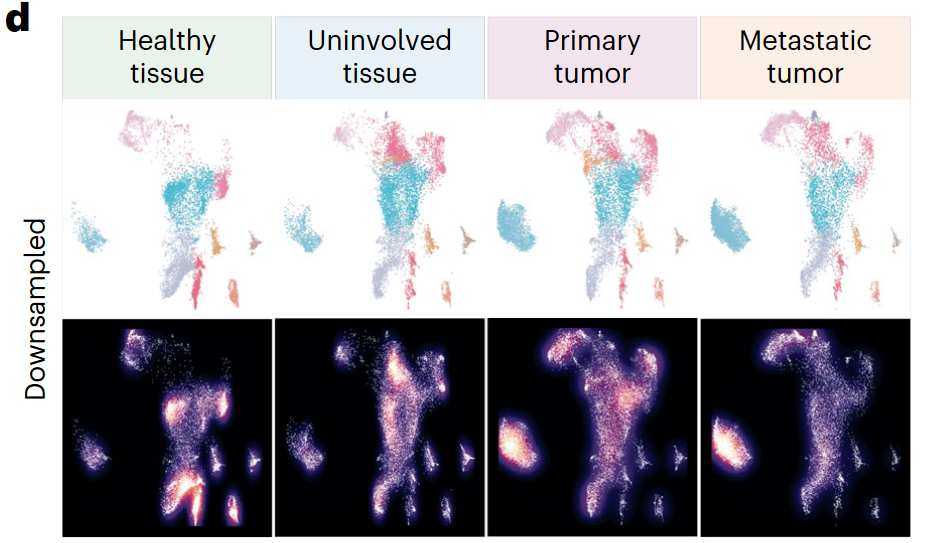
|
|
