DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy
Nucleic Acids Research | 2020 | IF=17

1、数据收集
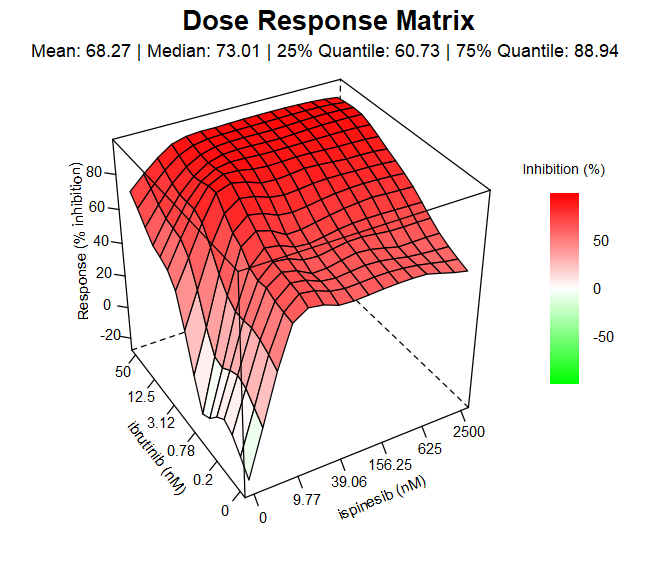
(1)HTS(high-throughput screening)高通量筛选技术可用于快速测得药物作用于癌症细胞系的不同剂量(dose concentrations)下的反应(Response)。其中,反应(Response)的指标常是细胞活力(cell viability)。
在细胞群体中总有一些因各种原因而死亡的细胞,总细胞中活细胞所占的百分比叫做细胞活力.
(2)药物组合测试(Combination Test)在应用HTS技术中,多为双药组合测试,用于评价组合效果(synergy协同、additivity叠加或者antagonism拮抗)。在具体实验中,通常设计n×n 剂量-响应矩阵(dose–response matrix)。
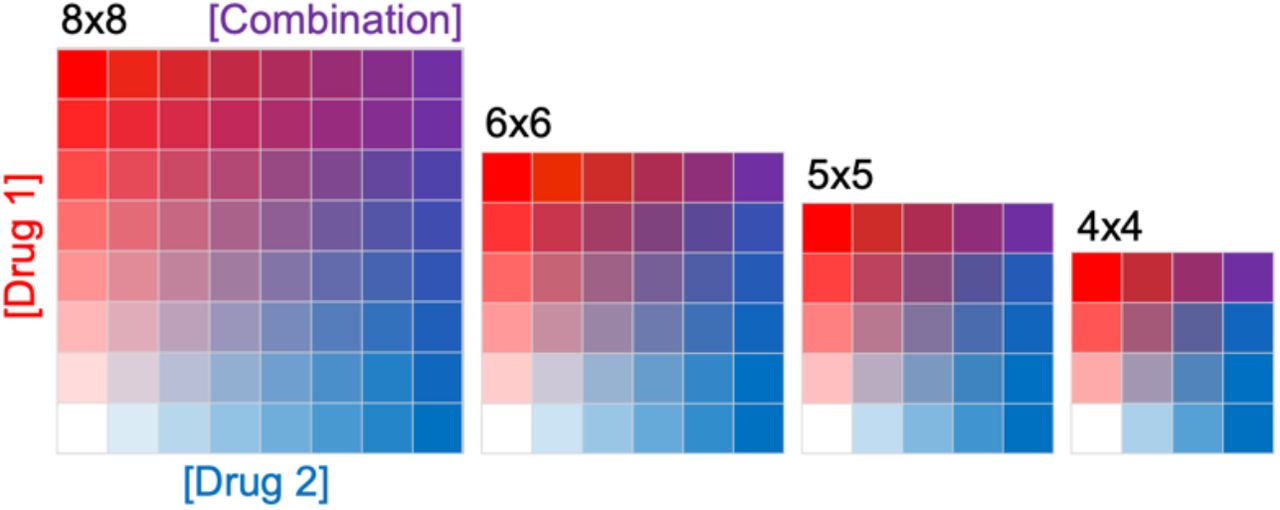
本文收集了大规模HTS assay 药物组合测试(Combination Test)实验结果。包括如下两个主要的来源。
-
NIH(National In-stitutes of Health)
-
主要收集自NIH下的NCI-ALMANAC (A Large Matrix of Anti- Neoplastic Agent Combinations)计划
-
311604个作用于60种肿瘤细胞系的药物组合3×3剂量反应矩阵,共2873514次test
-
-
文献:An unbiasedoncology compound screen to identify novel combination strategies.
- 92208个作用于39种肿瘤细胞系的药物组合4×4剂量反应矩阵,共1475328次test
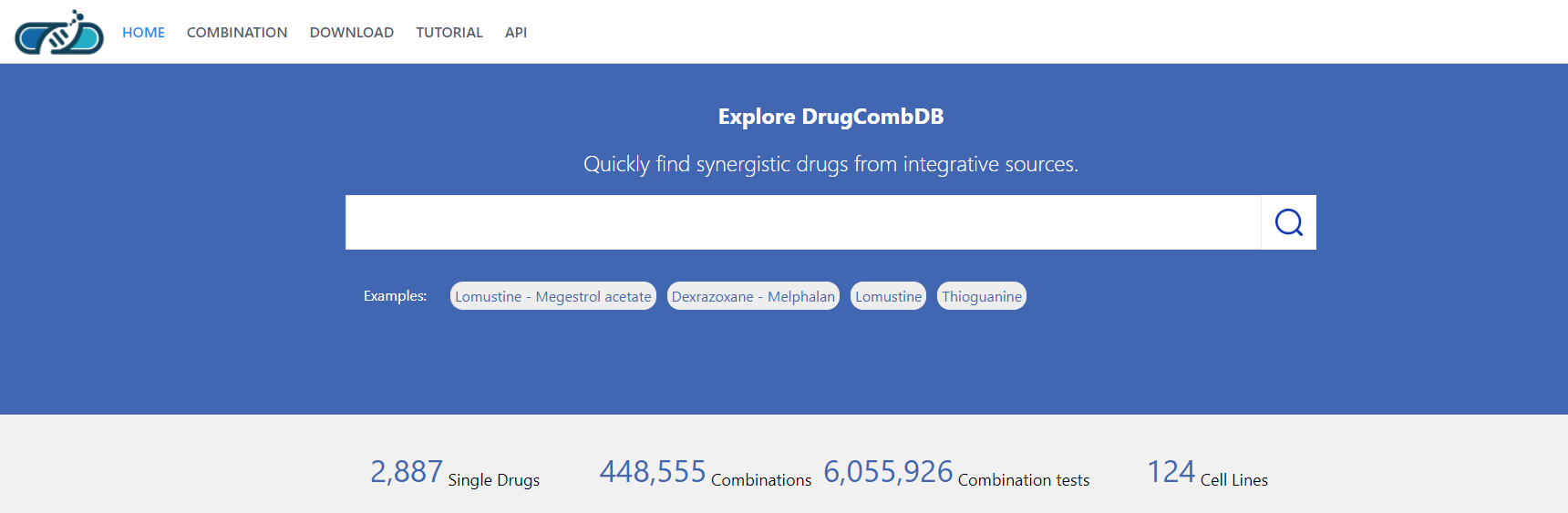
如下图所示,在文章发表时,根据作者说明已收集了 6055 92个combination tests,涉及448 555个双药物组合,2887种药物,以及124种肿瘤细胞系。
-
其它来源包括
-
文献研究汇总

-
其它数据库
- DCDB、DrugCentral均收集自 FDA Orange Book数据
- TTD:72种药物协同组合
- ASDCD:抗菌药物组合
- DrugBank:临床报道具有拮抗效应的antagonistic
-
文章提到,由于>=3药物组合的研究复杂性,在HTS实验少有涉及。在其它来源的收集中,也会注重区分出这些组合。
2、组合评价
通过药物组合的剂量-响应矩阵结果,去评价两药物间是否具有协同作用(synergistic)。
计算思路主要是:根据单药物的剂量-响应结果,得出双药物组合的**预期(expected)**剂量-响应。
- 如果实验结果优于预期响应值,则认为在该剂量组合下具有协同作用;
- 相反,如果低于预期值,则可能二者间具有拮抗(antagonistic)效果。
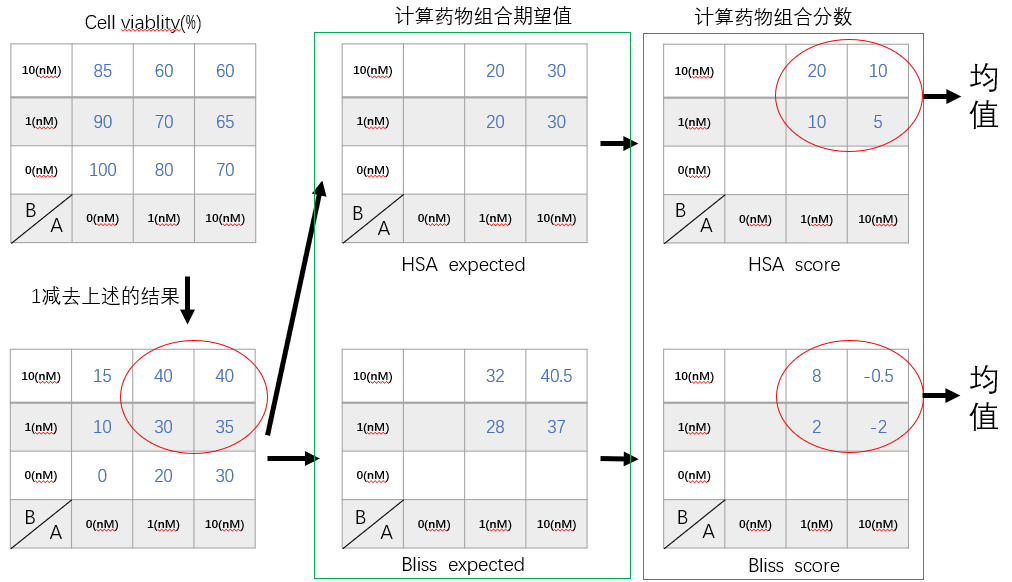
计算步骤(参考下图)
-
(1)首先计算结果(细胞活性viability)为0~1之间的百分数,为方便起见,常取百分值。
- 该值越大表明药物可增强细胞活力,反之则表示产生抑制效果。
-
(2)然后用1减去这些值,转换为药物的效果,结果越大(>0)表明抑制作用越强。
-
(3)计算药物组合的预期效果值,不同指标有不同的计算方式。
- 本文使用
SynergyFinderR包计算了HSA、Bliss、Loewe、ZIP4个指标 - 如下以HSA、Bliss两个指标为例演示
- 本文使用
-
(4)药物组合实际抑制效果减去预期抑制效果(如下公式)。
- Eab表示药物AB特定浓度组合的效果、Ea/Eb表示相同浓度下,单独药物的效果。(注意是百分数);
- 计算得到的Score值越大(大于0),表示该药物浓度组合越具有潜在的协同关系。
-
(5)最后取所有浓度组合的Score均值作为最终评价该药物组合的指标值。
$$ E_{HSA} = E_{AB} - max(E_A, E_B) $$
$$ E_{Bliss} = E_{AB} - (E_A + E_B-E_A×E_B) $$
3、整合数据
(1)对于不同HTS平台来源的药物组合实验结果(cell viability)具有可比性,作者对每个平台数据进行最大-最小归一标准化。 $$ R_{inhibit} = \frac{max(viability)-viability}{max(viability)-min(viability)} $$ (2)由于可根据Synergy score的正负性,简单判断两药组合是协同还是抑制关系。
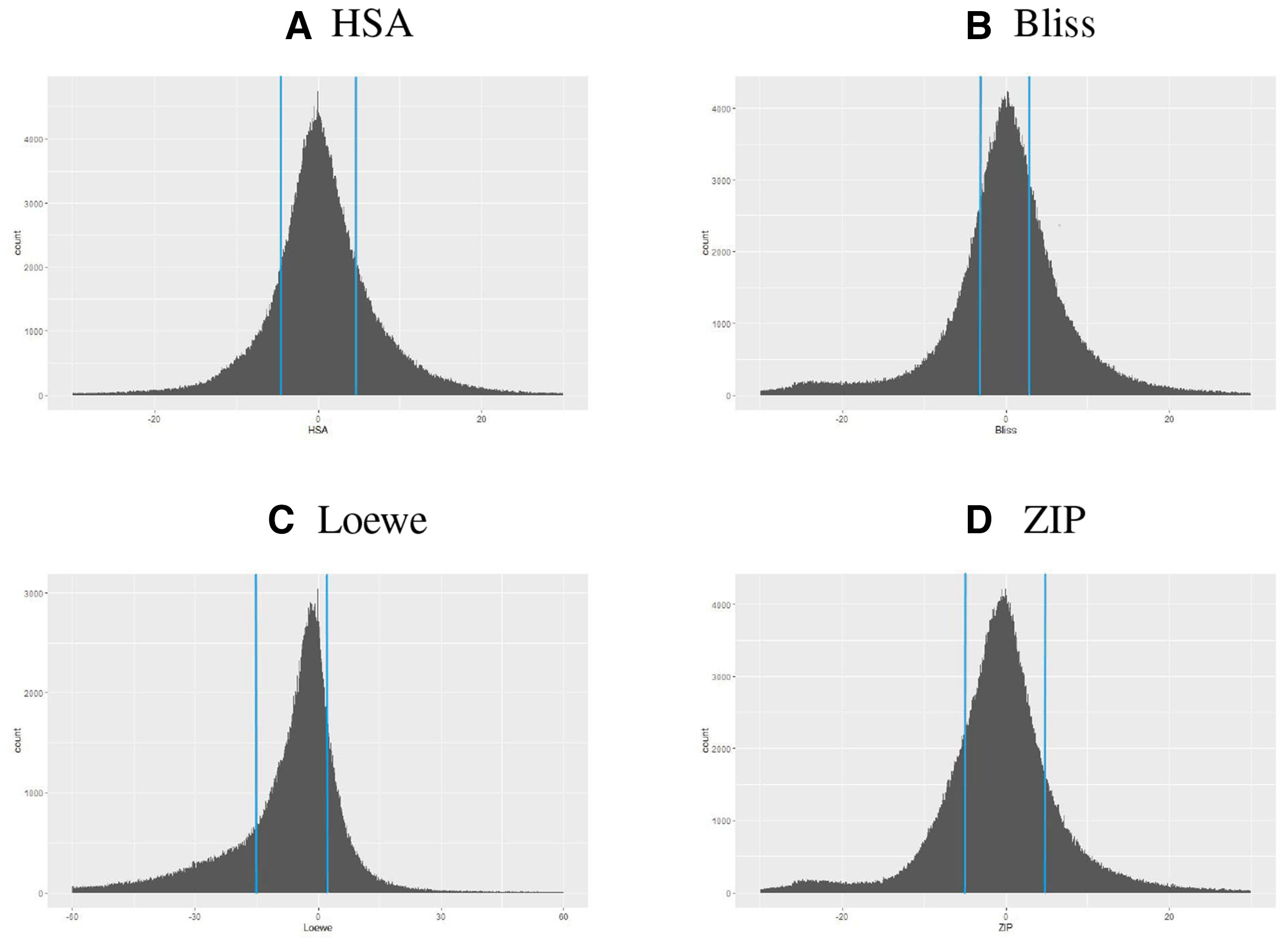
- 如下图所示,由于整体呈围绕0的正态分布,文章根据每种指标的Top25%,定义为synergistic;Low25%定义为 antagonistic
- 如果对于每一药物组合的4个指标都认为是协同(Top25),则认为该组合具有协同关系
- 最终根据上述过程,定义了85154个具有协同关系的两药组合;155824个具有拮抗关系的两药组合
(3)最后文章将收集整理的数据放到了自建的DrugCombDB网站里,供用户查阅、下载
最后数据库提供的化合物注释难以使用,后根据化合物名字在https://pubchem.ncbi.nlm.nih.gov/idexchange/idexchange.cgi网页进行化合物ID注释。
4、SynergyFinderR包使用
如上所说,文章使用了SynergyFinderR药物组合剂量反应矩阵的四种协同分数。下面简单学习这个包的用法。
|
|
4.1 输入数据
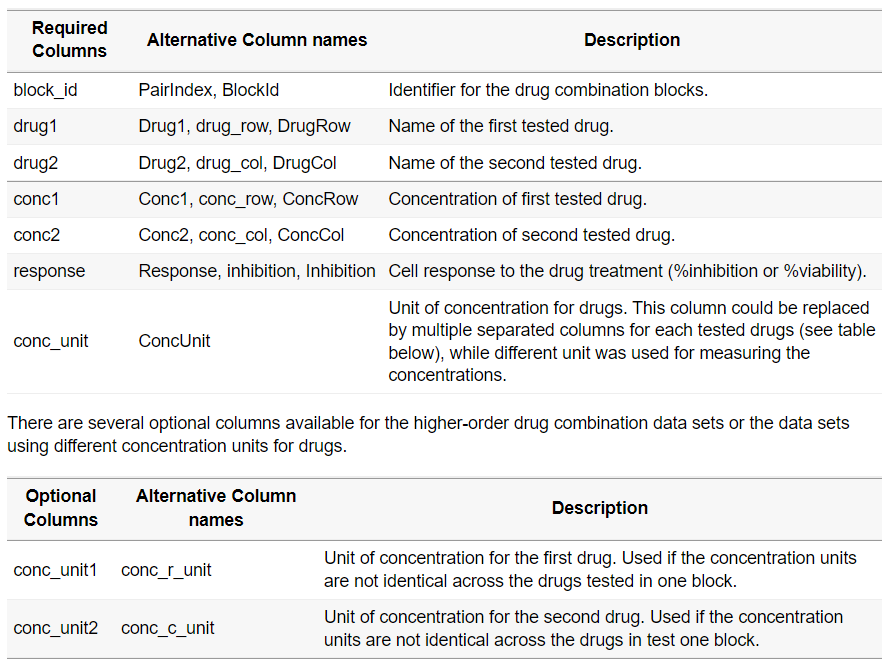
输入数据为一个表格,记录药物组合的相关实验信息;
SynergyFinderR对表格的列名具有严格的要求,具体如下
|
|
4.2 计算协同分数
- (1) 导入数据
|
|
- (2)计算协同分数
|
|
1、对于cell viability的response_origin实验结果,以及response理论上应该在(0,1)范围之间。如果不是,则可能属于异常值,可通过设置correct_baseline参数进行校正。具体可参考原文档。
2、这个包还可以计算IC50值,就不记录了。具体可参考原文档。
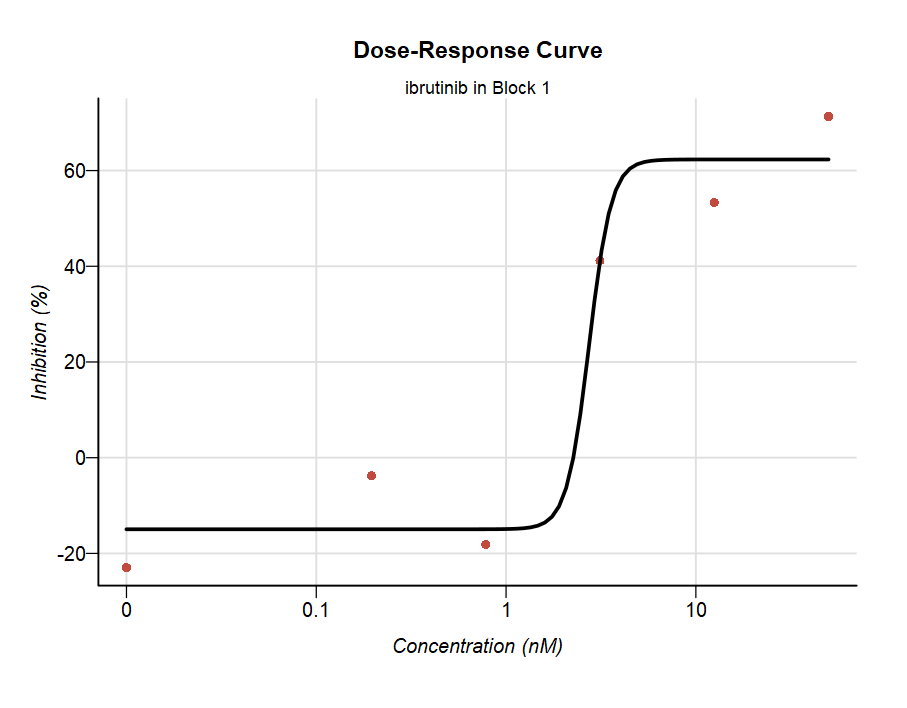
- (3)结果可视化
|
|
|
|
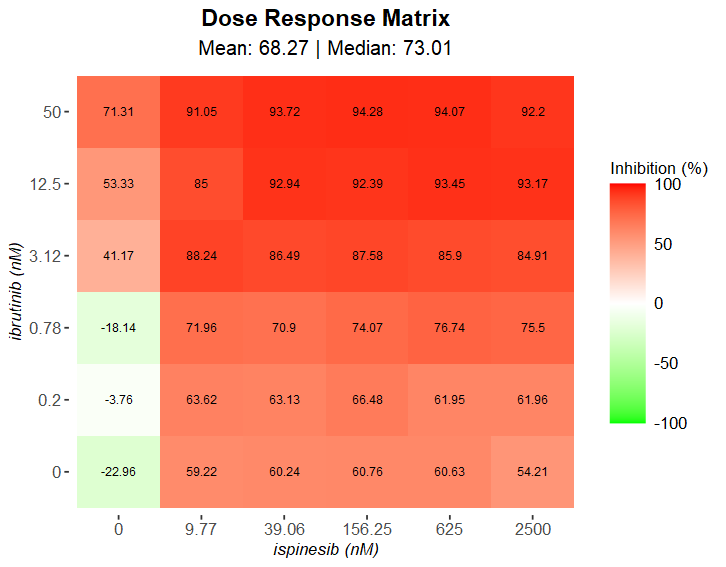
|
|
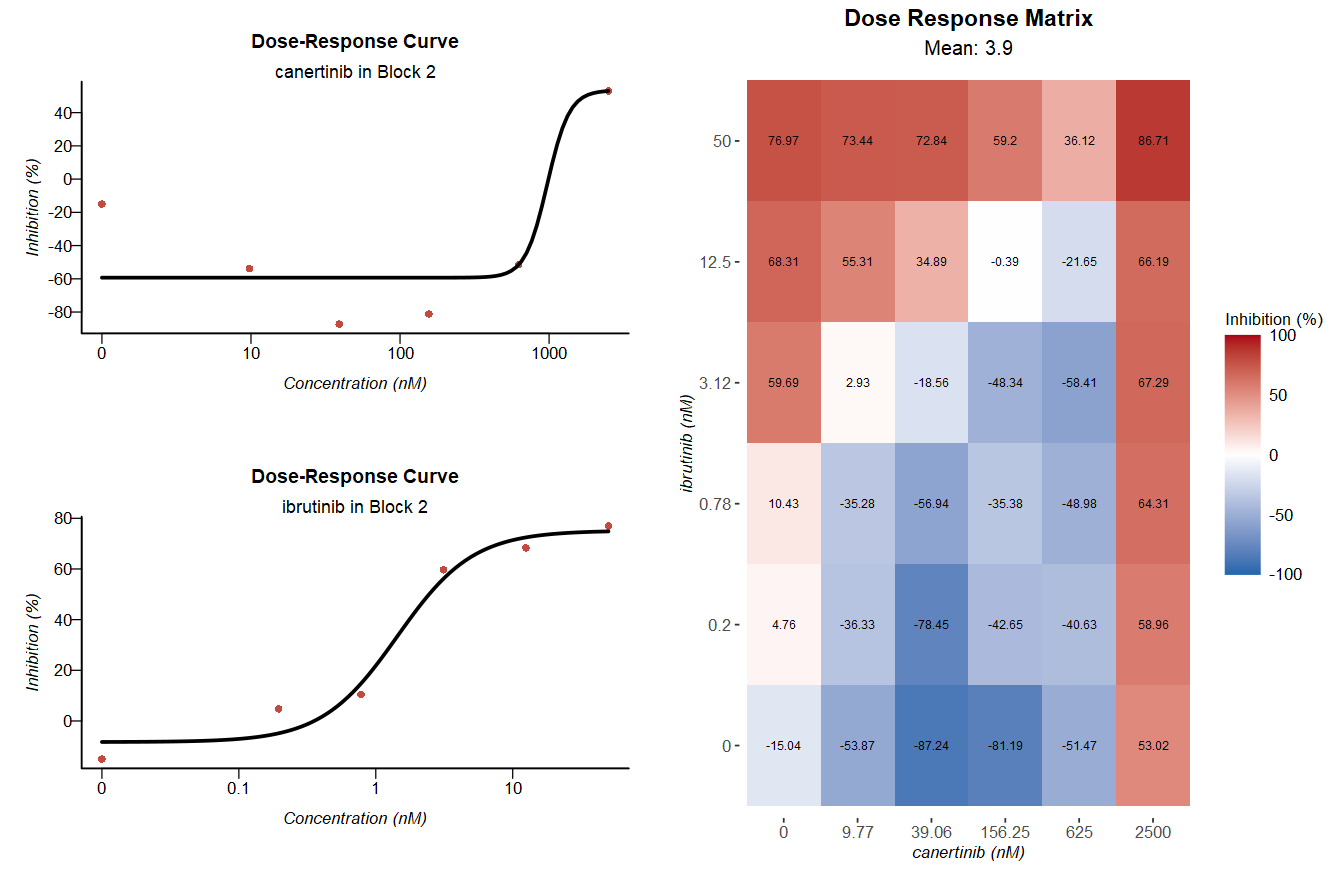
|
|