文章题目:M2-like tumor-associated macrophage-related biomarkers to construct a novel prognostic signature, reveal the immune landscape, and screen drugs in hepatocellular carcinoma
发表期刊及日期:Front Immunol, 2022 Sep 13

简单背景(wikipedia)—
巨噬细胞极化是一个巨噬细胞对应微环境讯号所表现不同程式功能的过程[1]。巨噬细胞极化有多种功能型态,他们可以完全极化成特定的表型,像是M1(典型活化巨噬细胞)或是M2(另类活化巨噬细胞)。
肿瘤相关巨噬细胞:(Tumor-associated macrophages,TAMs)是属于巨噬细胞细胞系的一种细胞,发现于肿瘤组织附近。TAM由淋巴循环中的单核细胞或组织中残留的巨噬细胞衍生而来,是浸润许多种肿瘤基质的白细胞的主要类型。

本文提及的M2-like tumor-associated macrophage如下简称为M2-TAM
0、文章小结
这篇文章的分析主要分为两个部分(1)结合传统转录组数据与单细胞转录组数据鉴定出与HCC M2-TAM高度相关的signature;(2)使用多种分析角度signature的重要意义,包括预后准确性分析、泛化能力验证、指导药物发现等方面。
这应该算是一篇常规的肿瘤预后类生信挖掘文章了—以signature为核心,桥接前(发现)后(论证)两大步骤,辅助以多种多样的分析策略。在本文中,单细胞数据仅出现在发现signature的步骤中。
1、简要步骤
结合如下流程图,文章分析步骤主要包括如下4步: (1)由Bulk RNA-seq的WGCNA M2相关模块与scRNA-seq的TAM细胞类型marker得到M2-TAM genes;
(2)从M2-TAM genes使用LASSO鉴定出最相关的signatures,并以通路富集分析、病人分群的生存分析等
(3)基于signatures计算的risk score将病人分为high/low group,进行生存分析、ROC分析,并以测试集验证;
(4)risk score与免疫浸润、免疫基因等的关系,并为相关药物发现提供参考依据。
2、结果概述
- 2.1 Bulk RNAseq数据发现M2相关模块
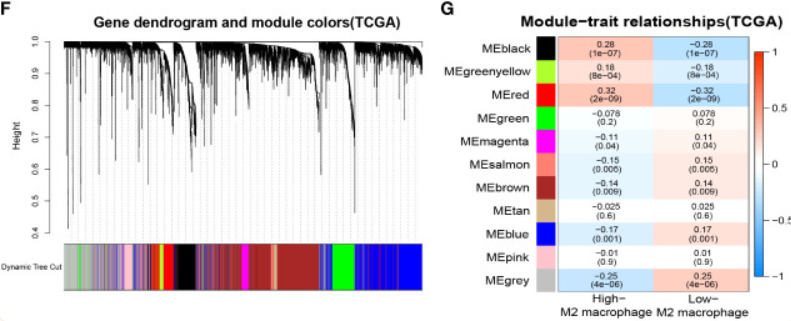
(1)对来自TCGA的HCC转录组数据分别进行WGCNA分析与CIBERSORTx免疫浸润分析
(2)寻找与样本M2细胞比例最相关的基因模块→包含405个基因的red module(correlation = 0.32, P<0.001)
- 2.2 单细胞数据鉴定TAM的高表达基因
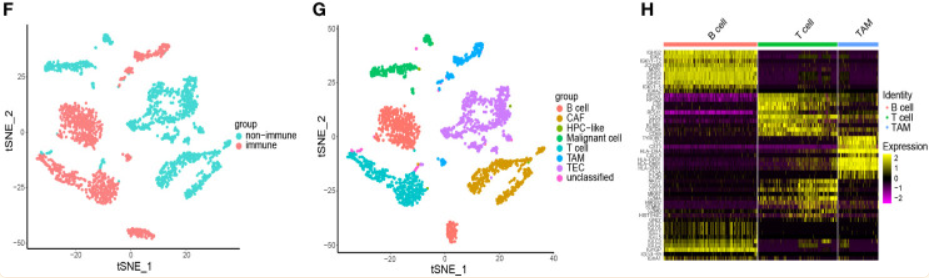
(1)使用GEO收集的HCC单细胞表达数据,进行质控、细胞类型注释(singleR)
(2)共得到包括TAM在内的3类免疫细胞,并分析得到TAM的2047个高表达基因
- 2.3 筛选得到最终的HCC相关M2-TAM基因
(1)取2.1得到的M2模块与2.2得到的TAM高表达基因交集,共127个基因;
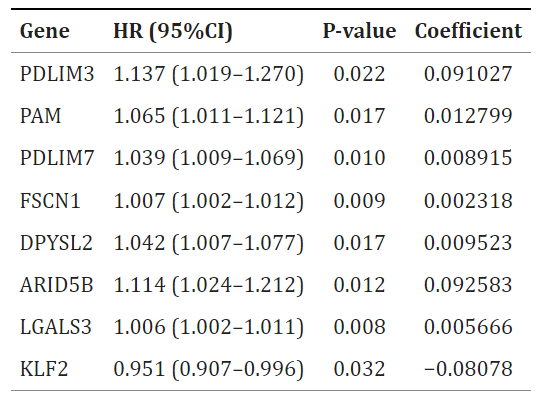
(2)先后进行单变量Cox回归与LASSO分析,得到8个基因与HCC预后高度相关的signature;
(3)对signature组成基因进行GO通路注释;
(4)使用signature组成基因对于病人进行分群,并以生存分析。
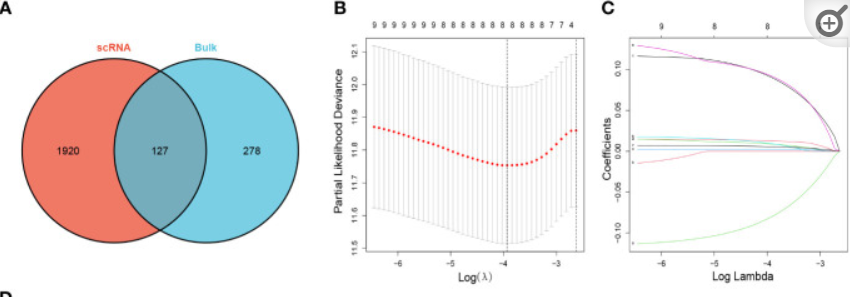
- 2.4 基于signature计算风险因子及衍生分析
(1)使用上述8基因的lasso系数为每个病人计算风险因子,并分为高、低风险组;
(2)结合病人的风险因子分数与高低风险组信息,进行生存分析、ROC分析、诺模图分析、GSEA分析等
(3)在收集自GEO的HCC转录组数据验证上述signature的预后泛化能力
- 2.4 基于risk score的深入分析
(1)risk score在不同临床特征亚类样本内部的可区分性(是否均显示high risk, worse prognostic)
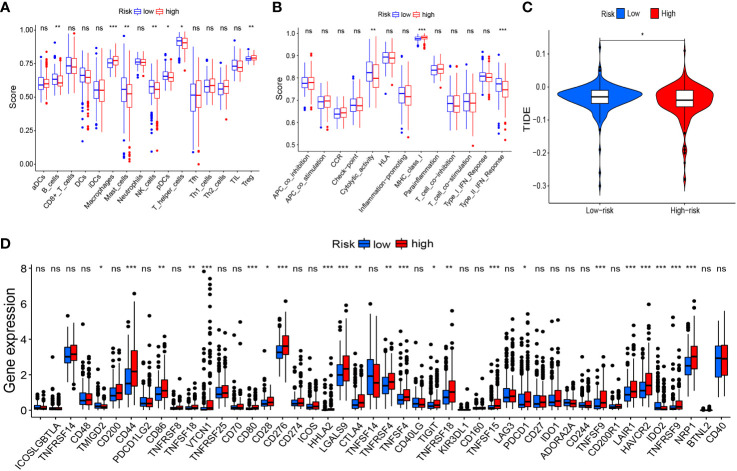
(2)risk score与样本的免疫浸润、免疫检查点相关基因表达程度的相关性(如下图)
(3)risk score与样本预测的化合物IC50(“pRRophitic” package)用于发现相关药物(化合物是否对于high risk具有更低的IC50)