1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
|
## 1) 两个最重要的参数 (后同)
# gene_list
gene_list="tests/data/gene_list.txt" # file path, one gene per row
gene_list= ["RAB2A","PRKAB2","PRKAA1"] # list of genes
# gene_sets (from enrichr API)
gene_sets='KEGG_2016'
gene_sets=['KEGG_2021_Human','GO_Biological_Process_2025']
gene_list = pd.read_csv("tests/data/gene_list.txt", header=None)[0].tolist()
gene_list[:4]
# ['IGKV4-1', 'CD55', 'IGKC', 'PPFIBP1']
enr = gp.enrichr(gene_list=gene_list, # or "./tests/data/gene_list.txt",
gene_sets=['GO_Biological_Process_2025'],
organism='human',
outdir=None,
)
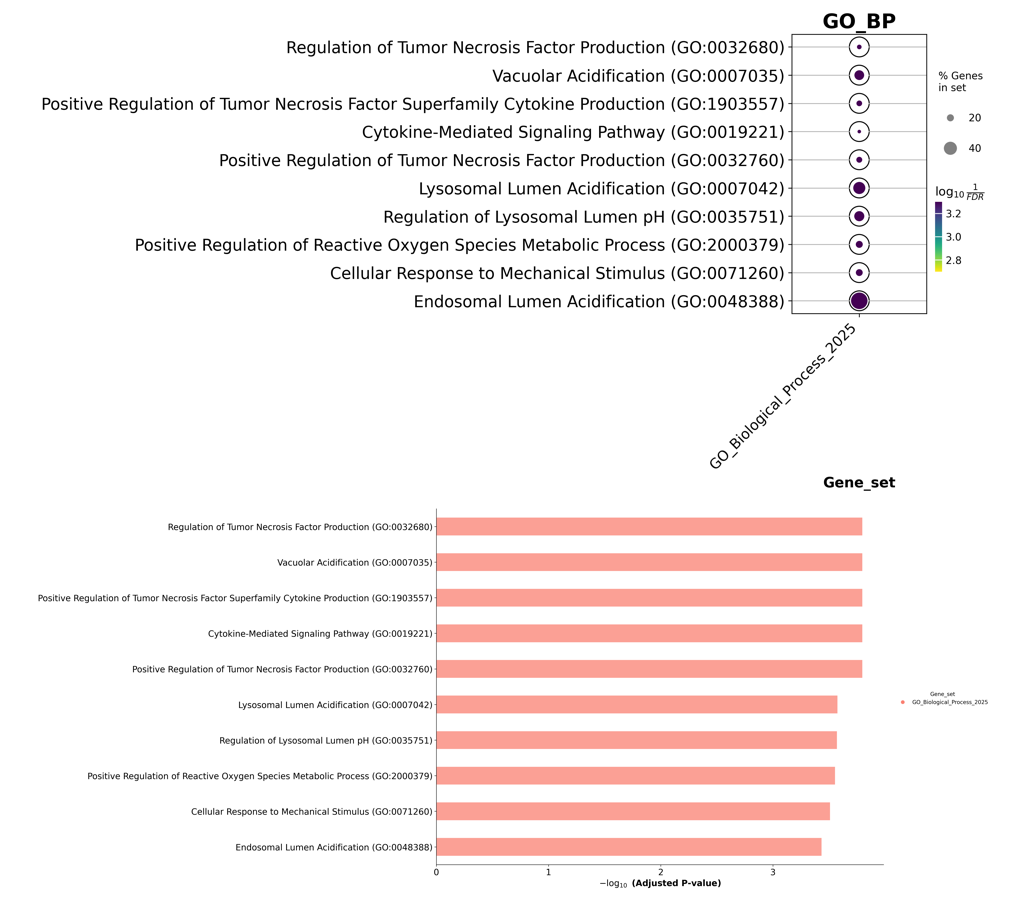
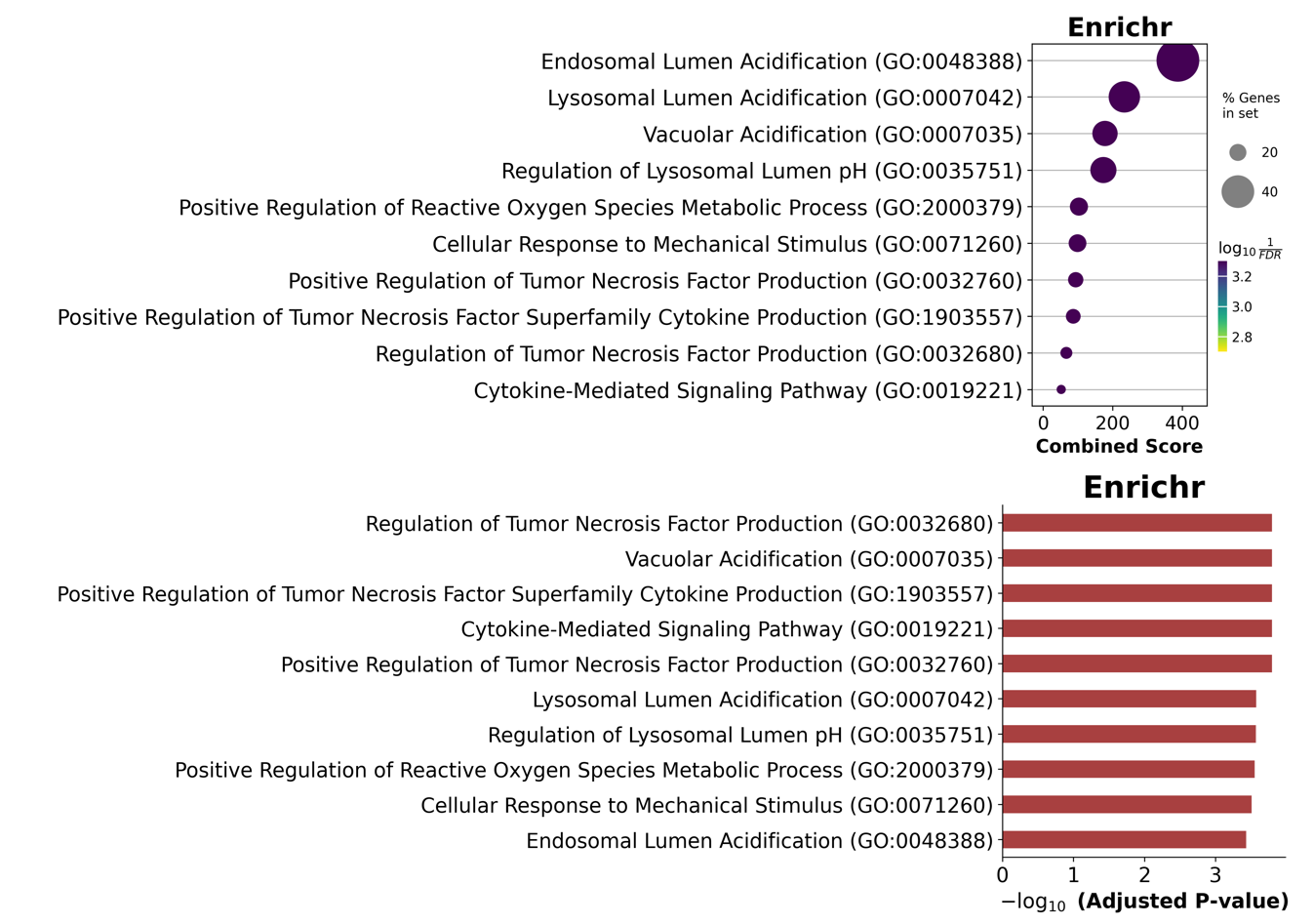
enr.results.head(5)
# Index(['Gene_set', 'Term', 'Overlap', 'P-value', 'Adjusted P-value',
# 'Old P-value', 'Old Adjusted P-value', 'Odds Ratio', 'Combined Score',
# 'Genes'],
# dtype='object')
## 2) 设置结果导出路径
enr = gp.enrichr(gene_list=gene_list,
gene_sets=['GO_Biological_Process_2025'],
organism='human',
outdir="enrichr_kegg",
)
os.listdir("enrichr_kegg")
# ['GO_Biological_Process_2025.human.enrichr.reports.txt',
# 'GO_Biological_Process_2025.human.enrichr.reports.pdf']
## 3) 自定义Background genes (P值会存在一定差异)
enr = gp.enrichr(gene_list=gene_list,
gene_sets=['GO_Biological_Process_2025'],
organism='human',
background="tests/data/background.txt", # 自定义背景基因 / or list object
outdir=None,
)
|